Data integrity is an old concept made essential in today’s digital age, and means data that is accurate, complete, and repeatable, which in turn ensures the product’s quality and public safety. In recent years, infractions relating to data integrity have been noted in several Food and Drug Administration (FDA) warning letters. The importance of record-keeping in drug manufacturing can be seen as far back as 1938, when the Federal Food, Drug, and Cosmetic (FDC) Act required the safety of new drugs be documented before being sold to the public, with similar regulations instigated in Europe and Japan throughout the 20th century.1
Production systems have large, inherent operational risks and are difficult to validate. Instead of exclusively reacting to public health disasters, preventative measures — such as the requirement for proof of claims — are taken to lower their likelihood and instill confidence in manufacturers.
All images: Particle Measuring Systems
Presence in regulations
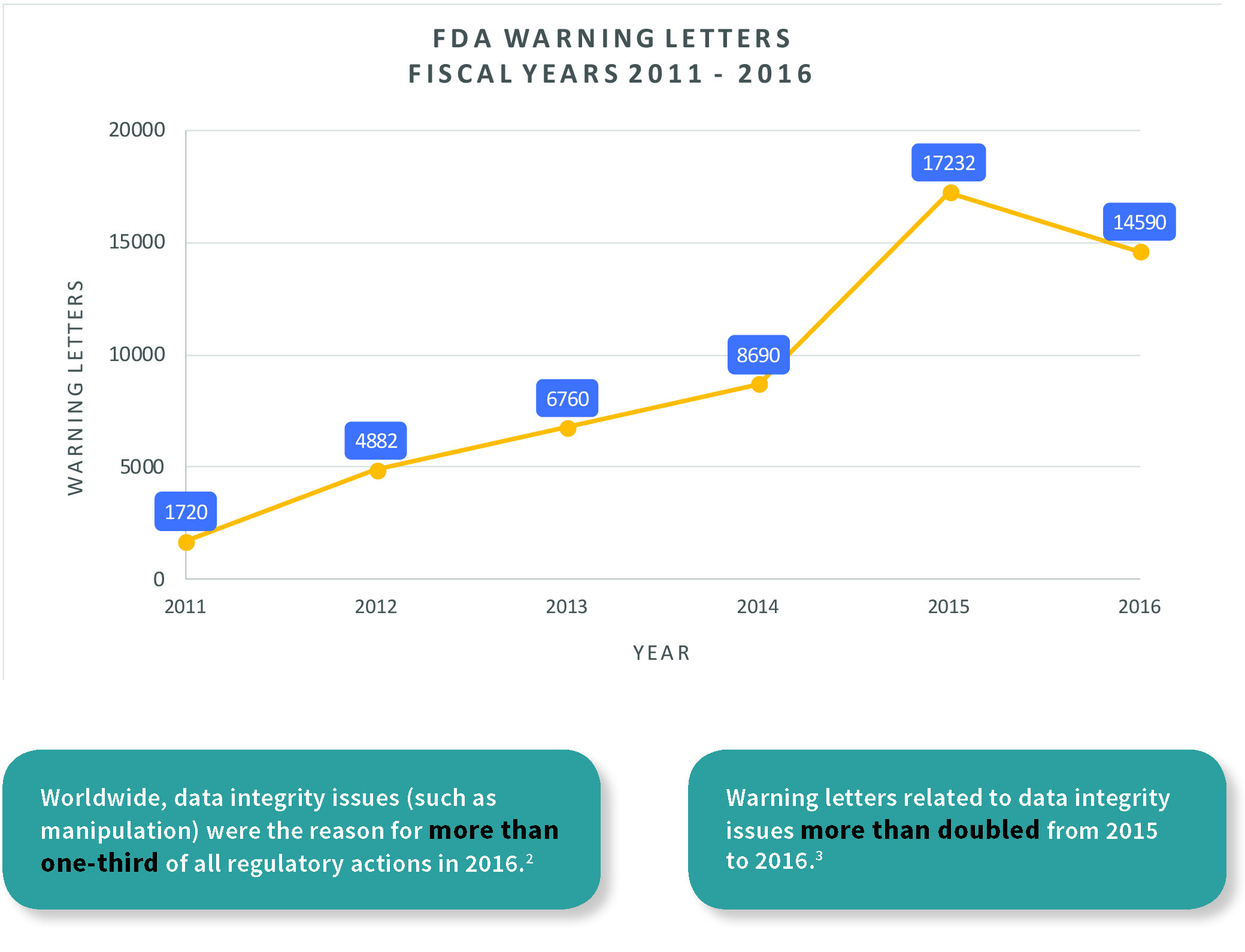
In the past few years, several FDA warning letters (483s) have been issued for data integrity deficiencies in the pharmaceutical industry. In 2016, more than 50 percent of MHRA warning letters involved data integrity lapses for computerized systems compared to the previous year.4 Inspectors are actively trained in data integrity requirements, and strongly enforce them for falsified batch records or discharging of raw data. Understanding recent standards, guides, and regulations pertaining to data integrity is essential to becoming compliant. Relevant documents include 21 CFR Part 11, MHRA: GxP, EU GMP Annex 1, the FDA Data Integrity and Compliance with cGMP, and the WHO Good Data and Record Management Practices. Inherent to data integrity compliance is the goal of increasing product quality, regulator confidence, brand reputation, and process control, while reducing product defects and costs. This applies to multiple areas of the pharmaceutical industry, including manufacturers of finished drug products for clinical trials, bioequivalence studies and commercial distribution, laboratories, contract manufacturing, suppliers, etc.
21 CFR Part 11
Title 21 of the FDA’s Code of Federal Regulations (CFR) Part 115 is the most widely used standard for appropriate data management. Part 11 applies to records in electronic format that are created, modified,
maintained, archived, retrieved, or transmitted according to requirements set in FDA regulations. Electronic records/signatures that meet Part 11 requirements may be used in lieu of paper records.
The document is divided into three parts:
General Provisions: The General Provisions section gives an overview of the terminology used throughout the document and the types of records that apply and do not apply.
Electronic Records: Signed electronic records shall contain:
• Signer’s printed name
• Date and time when the signature was executed
• Signature’s meaning (such as review, approval, responsibility, or authorship)
Computerized systems must be validated to ensure data accuracy, reliability, and consistency, and must be able to discern invalid or altered records. They also must generate an accurate, complete, and human-readable copy of the records. Access must be limited with the use of an ID and password. Those who develop, maintain, or use electronic record/electronic signature systems must have the education, training, and experience to perform their assigned tasks, which is essential to guarantee data integrity.
Electronic Signatures: Users who intend to generate electronically/digitally signed records must have a controllable form of identification. This control extends to maintaining a unique password and ID code (username), periodic password checks/changes, and configuration of individual and/or group ID privileges (i.e. differentiation between operator, supervisor and administrator). For example, it should not be possible for operators to have database management rights.
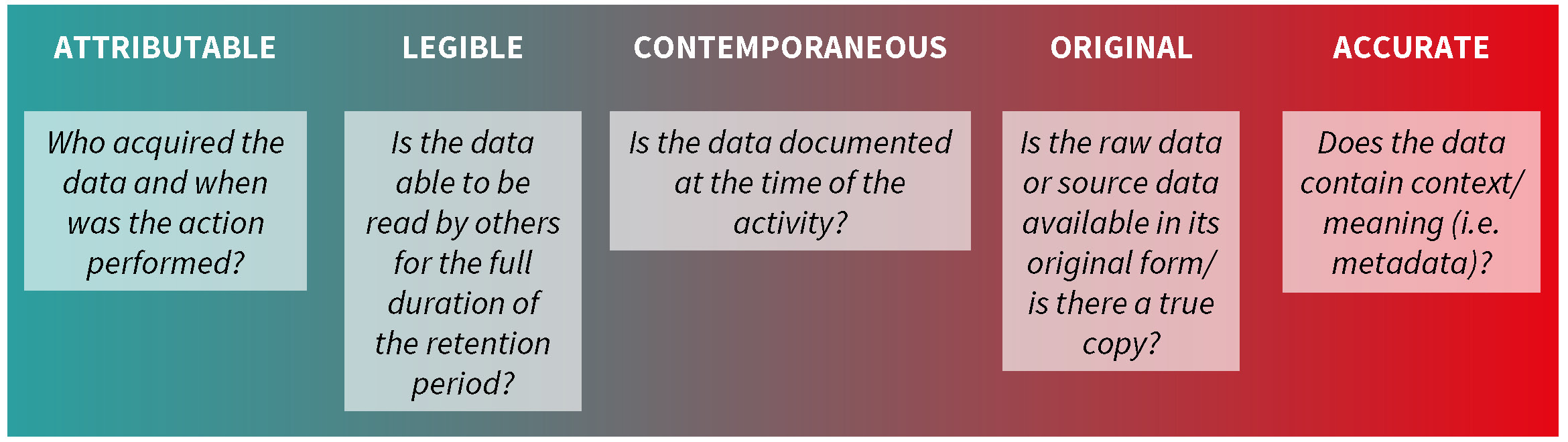

ALCOA
ALCOA is an acronym used by the FDA that stands for Attributable, Legible, Contemporaneous, Original, and Accurate.6 The concept behind ALCOA is that data quality directly impacts product quality, with focus placed on performing tasks correctly the first time and immediate reporting of results. As ALCOA is used in many FDA regulatory documents, it is important to be familiar with what is meant by each term.
EU GMP Annex 11
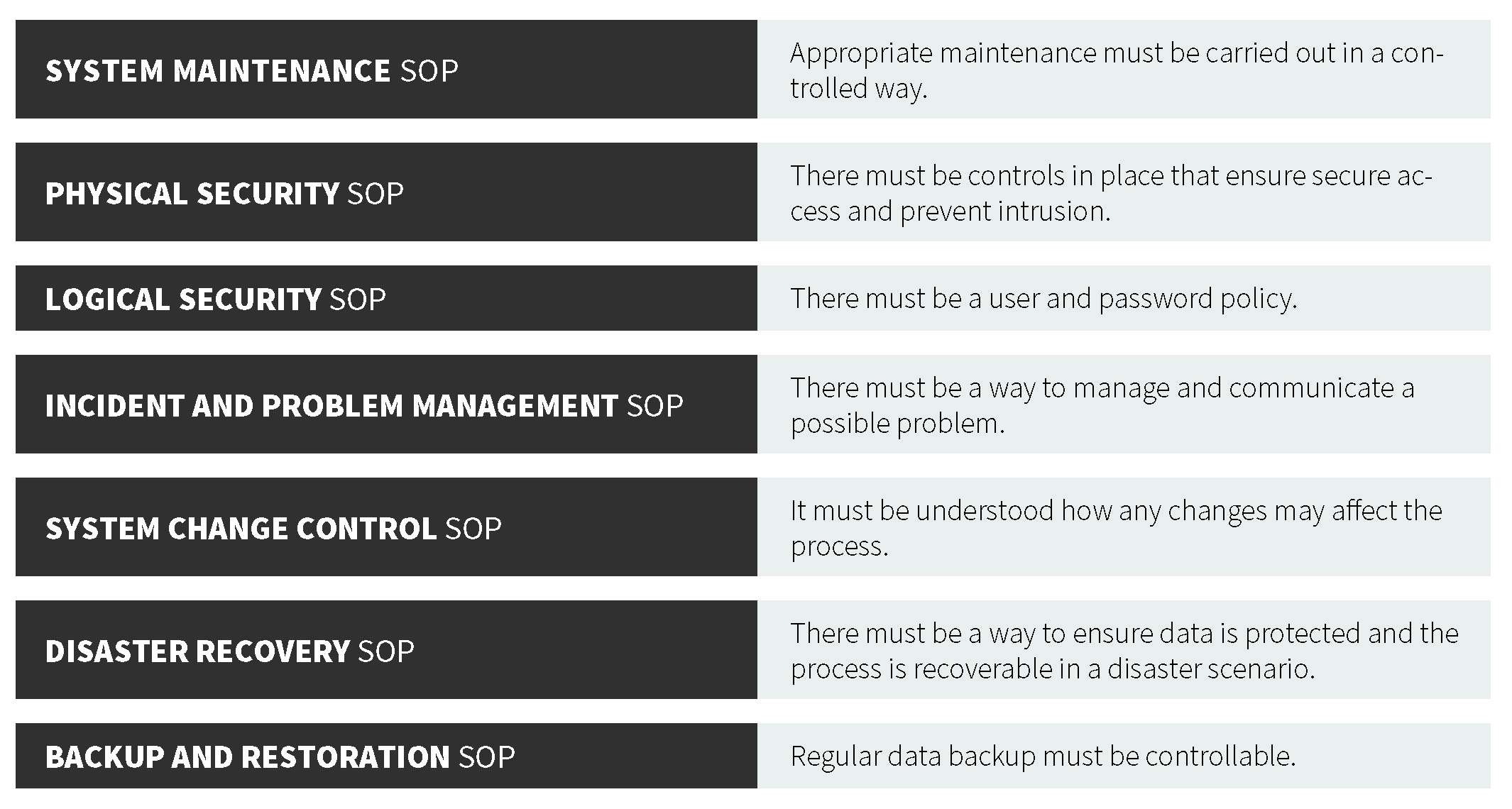
As part of the European Union (EU), EudraLex is the collection of rules and regulations governing medicinal products (for human and veterinary use). Annex 117 is part of the European GMP Guidelines and contains terms of reference for computerized systems used by organizations in the pharmaceutical industry. Note that Annex 11 is a guidance, not a regulation (21 CFR Part 11 is a regulation). Annex 11 defines the criteria for managing electronic records and signatures. These guidelines are similar to those of their U.S. counterpart. The central consideration of both the EU GMP Annex 11 and 21 CFR Part 11 documents is to ensure that records are entered correctly, cannot be tampered with, can be stored for the retention period as well as retrieved (in full) at any time during use and during the retention period. In each regulation, there is a strong focus on record accuracy, integrity, security, and retrieval. From the guidance, there are several important components of a proper data management system, which forms the basis for the following recommended standard operating procedures (SOPs).
FDA Data Integrity and Compliance with cGMP Guidance for Industry
This FDA guidance document is currently a draft guidance8 intended to clarify the role of data integrity in current good manufacturing practice (cGMP). The primary expectation is for data to be accurate and reliable. It does not establish legally enforceable responsibilities, but rather describes the FDA’s current thinking. It is not a required document to adhere to, but it is advisable to be familiar with the text. Key technical terms used in this document include:
• Static record format, where static indicates a fixed data document such as a paper record or electronic signature.
• Dynamic record format, which means a record that allows interaction between the user and record content, such as entering values manually in the system database.
• For a computer or related system, the “system” refers to the ANSI definition, which includes people, machines, and methods organized to accomplish a set of specific functions. In addition, this system can include compute hardware, software, peripherals, networks, cloud infrastructure, operators, and documents (manuals, SOPs, etc.)
MHRA GxP data integrity guidance
The MHRA GxP guidance document produced by the United Kingdom emphasizes that data integrity is fundamental to ensuring that medicines are of the required quality in a pharmaceutical quality system. The result of effective, robust data governance is complete, consistent, and accurate data used throughout the system.
Ethics
To safeguard against unethical behavior, the following is the recommended best practices:
• Data manipulation should not be allowed.
• All records should be created automatically (and not created after the fact from memory).
• Data should retain its unchanged time information.
• All pertinent data should be included in the record (even if it is undesirable).
• Passwords should not be shared between personnel.
• All original records should be stored and kept safely on non-volatile media.
• Records should never be discarded or destroyed.
Everyone involved in the process, no matter their position, is responsible for upholding data integrity. Data Integrity is a significant component of the Quality Management System, and inspectors around the world have made it very clear that good intentions are no defense against compromised data. The pharmaceutical industry must strongly consider any preventive or corrective action to improve product quality through an honest, ethical approach to data collection and retention.
References
1. FDA. (2006, June 30). The History of Drug Regulation in the United States.
2. PharmaCompass. (2017, January 19). 2016 — A Year of Data Integrity Issues and Pharma Non-Compliances. Retrieved from https://www.pharmacompass.com/radio-compass-blog/2016-a-year-of-data-integrity-issues-and-pharma-non-compliances
3. 2016 Warning Letters. (2016). Retrieved from https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2016/default.htm
4. MHRA. (2016, April). MHRA GMP Inspection Deficiency Data Trend 2016. Retrieved from https://mhrainspectorate.blog.gov.uk/2017/04/21/2016-gmp-inspection-deficiency-data-trend
5. FDA. (1997, March 20). Title 21 of the Code of Federal Regulations Part 11. Retrieved from https://www.ecfr.gov/cgi-bin/text-idx-?SID=86f097b6d76ecd7a285c372810e22a9f&mc=true&node=pt21.1.11&rgn=div5#se21.1.11_1300
6. South, G. (2018, February 26). Ensuring data integrity through ALCOA. Retrieved from https://www.pharmout.net/data-integrity-alcoa/#
7. European Commission, EudraLex. (2011, January). Annex 11: Computerised Systems. Retrieved from https://ec.europa.eu/health/documents/eudralex/vol-4_en
8. FDA. (2016, April). Data Integrity and Compliance with cGMP Guidance for Industry (Draft Guidance). Retrieved from https://www.fda.gov/downloads/drugs/guidances/ucm495891.pdf
Daniele Pandolfi is the Global Product Specialist, Aerosol, in Particle Measuring Systems’ Life Science Division. He has over 10 years’ experience in particle counter instrumentation and cleanroom contamination control, building close customer relationships. Daniele helps customers to solve their cGMP issues. www.pmeasuring.com



