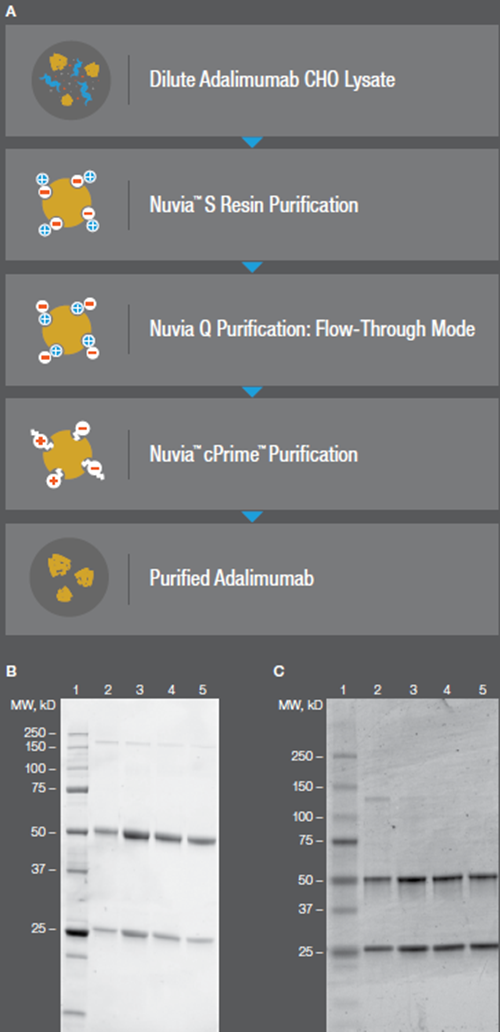
Figure 1. (A) Purification Strategy from CHO supernatant. Initial resin screening containing multiple resins (B) and later scaled up (C) showed comparable results
By 2020, the worldwide market for biologics is expected to exceed $390 billion (up from $46 billion in 2002) and account for 28% of the global pharmaceutical market. As they are developed across a wider range of therapeutic areas, biosimilars will play a more important role by providing a greater choice of treatment options.
A biologic is a therapeutic medicine manufactured using living cells. Up to $81 billion worth of currently approved biologics will have their patents expire by 2020 opening an opportunity for biosimilars to be developed as a cost-effective alternative. Bringing a biosimilar to market however, requires developers to demonstrate similarity to the reference originator biologic. This means that there are no significant differences between the two products. This information is provided as part of the totality of evidence which is reported to regulatory agencies.
Current Challenges
A number of significant challenges exist for manufacturers of biosimilars in the U.S. The FDA has not outlined what is required for a biosimilar to demonstrate interchangeability. Current guidance states that an interchangeable product can be expected to produce the same clinical result as the reference product and that there should be no increase in risk or decrease in efficacy. The benefit to the manufacturer is that the biosimilar could be automatically substituted for the reference product similar to how generics are substituted for their more expensive brand name counterparts. However, to date the FDA has not granted a designation of interchangeability to any of the approved biosimilars.
For biosimilar developers to produce a cost-effective product, identifying the best purification strategy from lab to process scale requires a reliable stream of workflow components from instruments to resins and columns. Costs must be controlled by designing a purification strategy with the least number of steps possible while retaining the purity, efficacy, and structure of the biosimilar.
This article presents a biosimilar workflow that addresses the challenges of developing a purification strategy, measures host cell protein and DNA impurities, outlines tools for comparability studies and characterization using biosimilar adalimumab, a HUMIRA-like biosimilar, as example.
Purification Strategy
The NGC Chromatography System and Nuvia resins from Bio-Rad were used to develop a three-step purification with reduced costs in comparison to a Protein A capture strategy. The modular NGC System can be configured to automate optimization and multicolumn or multidimensional (Multi-D) chromatography, which saves time and improves reproducibility when scaling up.
The rapid process development for biosimilar purification is depicted with Nuvia S Cation Exchange Resin for capture, followed by concentration, Nuvia Q Anion Resin in flow-through mode for intermediate purification, and Nuvia c-Prime Mixed-Mode Media used as the final polishing step (Figure 1).
Quantifying Impurities
The host cells used to generate biosimilars can be the source of protein and DNA contamination that may inadvertently affect the safety and efficacy of the therapeutic. Therefore, the concentration of these host cell proteins (HCPs) and host cell DNA (hcDNA) need to be reduced to levels that meet regulatory requirements.
Drug developers use antibodies to identify, detect, and quantify HCPs. Evaluation and validation of these anti-HCP antibodies can be quickly and efficiently carried out in less than two days. The anti-HCP antibody workflow requires sample prep, followed by 2-D electrophoresis, then a transfer of the proteins to a blot, followed by western blotting with the anti-HCP antiserum. Antibodies that meet the criteria of detecting relevant impurities can then be used in HCP ELISA. For developing this Biosimilar Workflow an HCP Detection ELISA Kit was used prior to, during, and after the three-step purification. The overall HCP reduction for the plate and scale-up column purification for each of the three steps were >90%, >99%, and 99.99%.
Droplet Digital PCR (ddPCR), a sensitive and absolute way to quantify hcDNA, was used. This method eliminates the need for a DNA extraction step associated with the traditionally used qPCR method and allows for direct quantification. The ddPCR Host Cell Residual DNA Quantitation kit was used to measure picogram (pg) levels of hcDNA per milligram of adalimumab. From raw feedstock to the final purification hcDNA levels went from 2.84 x 10^11 pg to 5.3 pg.
Assessing Comparability of Biosimilars
Another part of the biosimilar workflow requires comparison studies confirming that biosimilars function similarly and are as efficacious as their originator biologic. Cell-based assays are often chosen to show comparability because of the wide range of information they provide.
To show adalimumab biosimilar’s comparability to HUMIRA, murine L929 cells treated with TNF-α only, TNF-α with the purified anti- TNF-α antibody adalimumab, and TNF-α with purchased control anti-TNF-α antibody adalimumab were analyzed by flow cytometry. The profile of live versus dead cells was compared between TNF-α with the purified and purchased control version of adalimumab. Both the purified and control adalimumab showed a comparable protective effect on the cells (Figure 2).
Characterization
Finally, there must be no clinically meaningful differences between the biosimilar and the reference product in terms of safety, efficacy, and immunogenicity. Antibodies to biotherapeutics can be used for detection of free or total drug, or drug bound to its target. These anti-drug antibodies (ADAs) can also be used as reference standards in assays developed to measure patient response to the drug. Using fully human ADAs eliminates the need to source antibodies raised in animals.
To meet regulatory requirements and to prove that the biosimilar has no clinically meaningful differences from its originator biologic, it is imperative that developers employ sensitive and reliable analytical methods. Having an ability to minimize the required amount of time spent in each development step as well as the associated costs involved in the process are also important. Using this simplified, sensitive, and reliable biosimilar workflow enables developers to attain their goal of gaining regulatory approval faster.
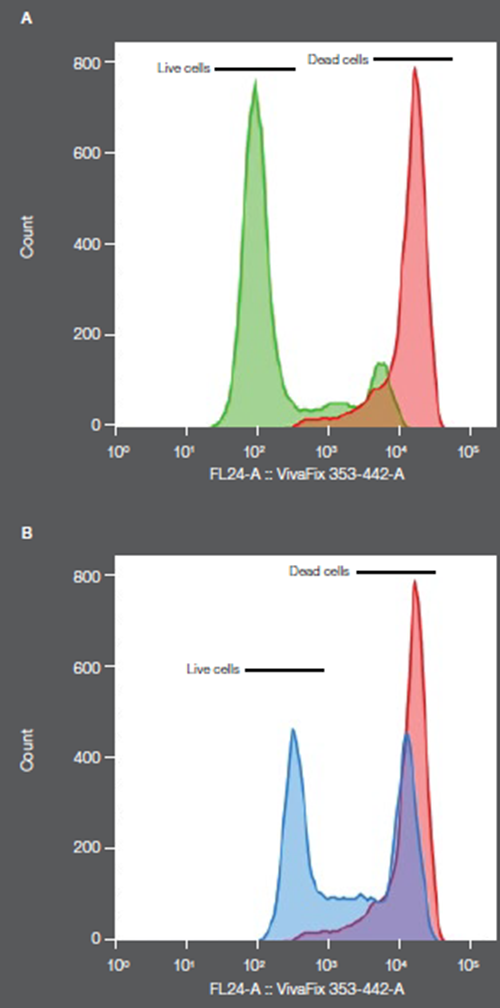
Figure 2. Comparability test of an Adalimumab biosimilar purified from lysate, in green, compared to commercial biologic, in blue. (In red: TNF-only treated cells)