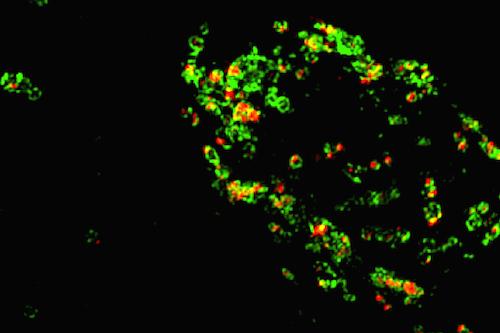
Red shows where granulins are in cells, and green is LAMP1, a marker of lysosomes. Source: Chris Holler, Emory University School of Medicine
Emory University School of Medicine researchers have developed tools that enable them to detect small proteins called granulins for the first time inside cells. Granulins are of interest to neuroscientists because mutations in the granulin gene cause frontotemporal dementia (FTD). However, the functions of granulins were previously unclear.
FTD is an incurable neurodegenerative disease and the most common type of dementia in people younger than 60. Genetic variants in the granulin gene are also a risk factor for Alzheimer’s disease and Parkinson’s disease, suggesting this discovery may have therapeutic potential for a broad spectrum of age-related neurodegenerative diseases.
The results were published August 9 by the journal eNeuro (open access).
Some neuroscientists believed that granulins were made outside cells, and even could be toxic under certain conditions. But with the newly identified tools, the Emory researchers can now see granulins inside cells within lysosomes, which are critical garbage disposal and recycling centers. The researchers now propose that granulins have important jobs in the lysosome that are necessary to maintain brain health, suppress neuroinflammation, and prevent neurodegeneration.
Problems with lysosomes appear in several neurodegenerative diseases such as Alzheimer’s and Parkinson’s.
“A lysosomal function for granulins is exciting and novel. We believe it may provide an explanation why decreased levels of granulins are linked to multiple neurodegenerative diseases, ranging from frontotemporal dementia to Alzheimer’s,” says senior author Thomas Kukar, PhD, assistant professor of pharmacology and neurology and the Emory University Center for Neurodegenerative Disease.
Granulins are derived from a larger precursor protein called progranulin. A genetic deficiency in progranulin explains some forms of frontotemporal dementia (FTD), both familial and sporadic.
If both copies of the progranulin gene are mutated, the result is a rare disorder: neuronal ceroid lipofuscinosis (NCL). NCL is a member of the larger group of lysosomal storage diseases (LSDs), inherited diseases caused by defects in lysosome function. NCL can be caused by mutations in several genes encoding lysosomal proteins. NCL is partly named after lipofuscins – deposits of fats and proteins – which accumulate inside cells because of impaired lysosomes. NCL typically appears earlier in life, with features such as blindness, developmental delay, motor problems and seizures.
“Over the past few years, evidence has accumulated that progranulin deficiency impairs the function of lysosomes, but scientists still don’t know why”, says assistant scientist Chris Holler, PhD, the first author of the paper.
“Although some people suspected that progranulin gets cleaved in the lysosome, it could not be proven,” he says. “Our paper is the first to actually show that mature granulins are made in the lysosome.”
“More importantly, the granulins are stable in lysosomes suggesting they have an important function, rather than just an inert by-product”, he adds. The next step is to use the antibody tools to probe what granulins are doing in lysosomes. Granulins may work together with similar lysosomal caretakers called saposins, Holler says.
The findings point to a strategy for treating diseases caused by progranulin deficiency (both FTD and NCL, possibly) by supplying replacement granulins, either as proteins or through gene therapy. The protein/enzyme replacement approach is already used for other forms of NCL and LSDs.



