This seventh and final installment in the Modernizing Chemistry Code series covers how scientists are updating popular molecular dynamics, quantum chemistry and quantum materials code to take advantage of hardware advances, such as the Intel Xeon Phi processor.
“In our group, we use computational chemistry methods to study physical and chemical processes in biochemical systems,” said Dr. Vladimir Mironov, Department of Chemistry, Lomonosov Moscow State University. “With the help of computational chemistry and HPC systems, we can conduct experiments to predict properties and biological activity of the systems. Without the aid of supercomputers and HPC software, experiments could be very expensive and/or time-consuming or even impossible.” While advances have been made in both hardware and software, much of the HPC software code is decades old. Changes are needed in HPC software so that it can take advantage of advanced hardware to let scientists do research faster and on more complex biological or chemical systems.
GAMESS (General Atomic and Molecular Electronic Structure System) is a multi-purpose computational chemistry code that has an estimated 150,000 users. The Gordon research team at Iowa State University, led by Dr. Mark Gordon, professor of chemistry, has maintained GAMESS US code for decades and spent countless hours optimizing the code to run on current architectures. “The Department of Energy and National Science Foundation (NSF) are focused on moving to the exascale. Electronic structure algorithms must take advantage of novel computer architectures, such as the interesting new architectures being provided by Intel,” said Gordon.
With such a large user base, GAMESS is used for just about every imaginable application in chemistry and biochemistry, and materials engineering, chemical engineering, and mechanical engineering. GAMESS is a general-purpose suite of electronic structure and quantum mechanics/molecular mechanics (QM/MM) methods that can be run on virtually any computer, cluster, massively parallel system, or for that matter a desktop Mac or PC.
GAMESS can perform a number of general computational chemistry calculations, including Hartree–Fock, density functional theory (DFT), generalized valence bond (GVB), and multi-configurational self-consistent field (MCSCF) energies and Hartree-Fock Hessians. GAMESS code also includes newer methods in quantum mechanics, including approaches for extrapolating to the full CI and complete basis set limits, novel methods for CASSCF calculations, new coupled cluster techniques, methods for evaluating non-adiabatic and relativistic interactions, new approaches for distributed parallel computing, and QM/MM methods for describing solvent effects and surface science. In addition, the Iowa State team developed a Distributed Data Interface (DDI) and a later generalized DDI (GDDI) that enables distributed parallel coding and multi-level parallelism.
History of GAMESS Code
GAMESS (General Atomic and Molecular Electronic Structure System) is a computational chemistry software package that was created from several existing quantum chemistry programs, particularly HONDO, by the staff of the National Resources for Computations in Chemistry (NRCC). The original creators of the program were Michel Dupuis, D. Spangler, and J. J. Wendoloski, as part of the old NSF National Resource for Computational Chemistry in the late 1970s
Funding of many of the developments in GAMESS from 1982 to the present time was provided by the Air Force Office of Scientific Research. The present version of the program has undergone many changes since the NRCC days. Michel Dupuis developed two programs GAMESS and HONDO. The two were essentially identical. Michel decided to focus on HONDO and the Gordon group took over the development of GAMESS in 1982, at North Dakota State University. GAMESS US code with the Gordon team was moved to Iowa State University with an increased emphasis on optimizing code for parallel processing. A team in the UK obtained a similar version and created GAMESS UK. The Gordon research group at Iowa State University currently maintains GAMESS (US) source code.
Scientific Case: Molecular Modeling of Human Enzymes
Moscow State’s modeling of the molecular polymorphism of human enzymes is an example of quantum chemistry scientific research being done with supercomputers. Humans differ from each other by their genetic code. It often predetermines the possibility of getting diseases (like cancer or Alzheimer’s disease) or sensitivity to drugs. The Moscow State team studies the role of harmful mutations in the proteins like butyrylcholinesterase (BChE), carboxylesterase (CES1) and small GTPases of the Ras family.
As an example, Ras is a protein that transfers the signal to the process of cell growth and division. In living cells, Ras acts as a molecular switch. Its signal transduction ability depends on its molecular bonding. When it is bound to the guanosinetriphosphate (GTP) molecule, the switch is turned “on.” When the bound molecule is guanosinediphosphate (GDP), the signal is inactive. The process of switching the protein off is controlled by a special GAP protein. Inside the Ras-GAP protein, the complex GTP molecule transforms to the GDP molecule. A malfunction of this process may result in uncontrollable cell division and finally to the formation of a tumor. According to various estimates, mutations of the Ras protein are observed in 16 percent to 30 percent of human tumor cases. Most of these cases are related to mutations of amino acids in the 12, 13 and 61 positions in the Ras peptide sequence.
“We use quantum chemistry calculations to understand the mechanism of GTP-GDP transformation inside the Ras-GAP protein complex and to find an explanation of why these amino acids are so important. According to our studies, an amino acid in position 61 actively participates in the reaction and its substitution would significantly decrease the GTP to GDP transformation rate. Amino acids in the 12 and 13 position are not directly involved into the reaction; however, the calculated slowdown of the transformation rate is in good agreement with the experimental data. “The calculated model of GTP-GDP transformation in Ras-GAP protein complex can be used in drug discovery for Ras-related tumors and also for the study of the mutations in similar proteins,” said Mironov.
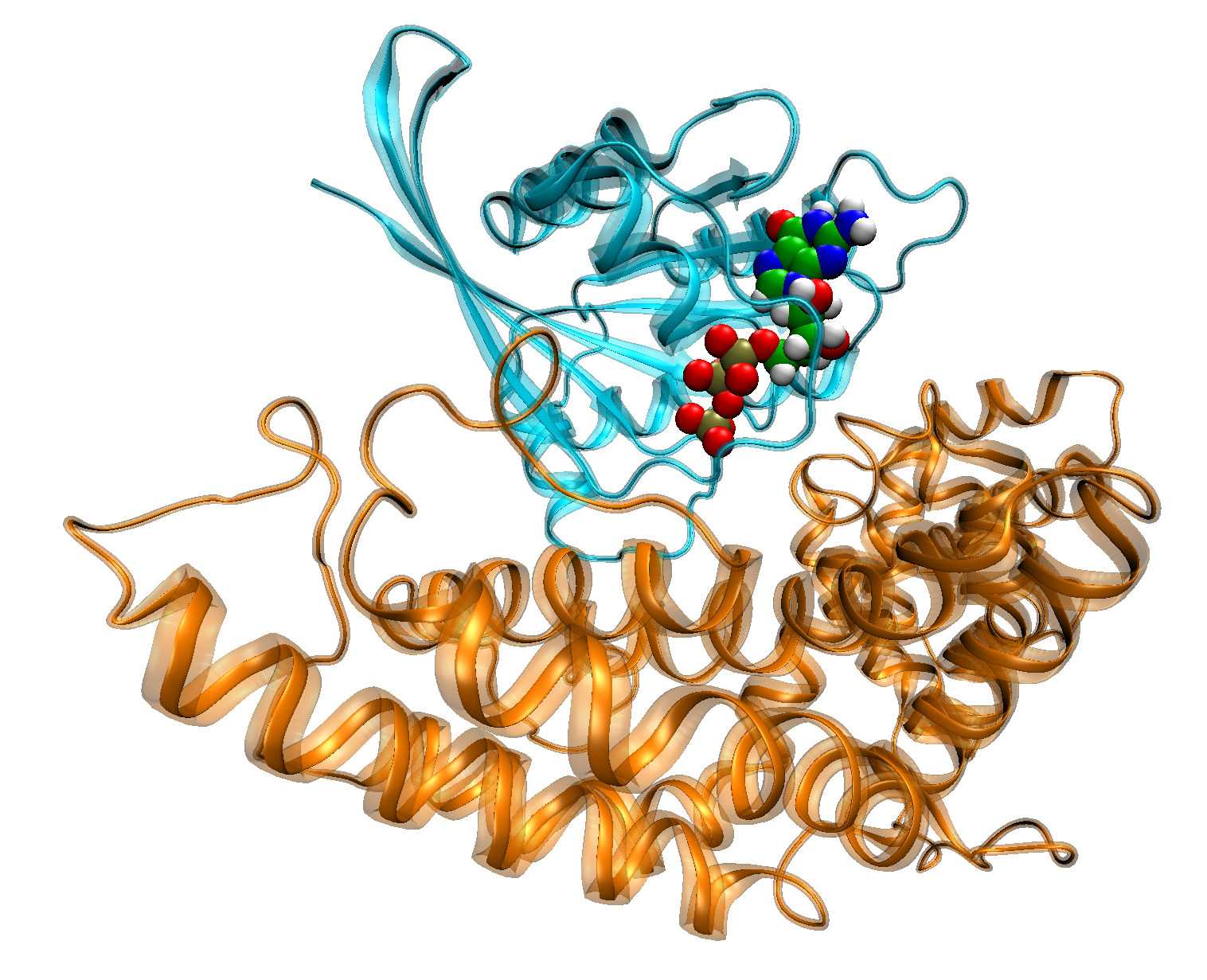
Figure 1: Ras-GAP protein complex with GTP molecule. Courtesy of Dr. Vladimir Mironov, Moscow State University.
Iowa State Maintains the GAMESS Code and Is Modernizing It For Current Hardware
The GAMESS developers at Iowa State University and Ames Laboratory are in the process of modernizing the code so it can use the features of the Intel Xeon Phi coprocessor. “Some parts of the GAMESS code (such as the integral code) are old, so our team is focusing on rewriting parts of the code which are particularly computationally demanding. The GAMESS code does not currently take advantage of threading, so a focus is rewriting parts of the code to be able to take advantage of the threading capabilities in Intel Xeon processors and particularly Intel Xeon Phi coprocessors. In optimizing the GAMESS US code, the Iowa State team addresses the performance of Hartree-Fock (HF) theory, density functional theory (DFT), second order perturbation theory (MP2) and coupled cluster theory (CCSD(T) and CR-CC(2,3)), for both closed and open shell species. The energy can be calculated for all of these systems and the energy gradient can be calculated for all but the coupled cluster methods. The calculations are complex and time consuming and there can be as many as 109 or 1010 floating point integrals.
Iowa State System
Cyence*: 297 nodes each with two 8-core Intel Xeon processors E5-2650
24 nodes with dual Intel Xeon Phi coprocessors 5110p, 24 nodes with dual NVIDIA* k20
Cyence* CPU clock speed 2.0 GHz
A smaller system in the Gordon lab uses the newest Intel processor code name Haswell CPUs. Another group system has dual Intel Xeon Phi 7120P coprocessors.
The Iowa State team uses Intel Xeon processors for the vast majority of their in-house computer clusters used for scientific applications. “We expect that our Intel IPCC efforts to vectorize and improve the performance for Intel Xeon Phi coprocessors will translate into even better performance on Intel Xeon processors. Research is underway to study the performance of various features of GAMESS on Intel processors and coprocessors.
There is a trade-off between time to solution and energy/power consumption. As part of an Air Force grant, the Iowa State group and colleagues have obtained two of the latest Intel Xeon Phi coprocessors, in order to study the effect of reducing the frequency on time to solution. Masha Sosonkina at Old Dominion University is leading this effort. In a recent paper, we demonstrated that considering both time to solution and energy/power consumption, the Intel Xeon Haswell system beats the ARM-64*. “This advantage may not last,” said Gordon.
Scientific Case: Silanol-Assisted Carbinolamine Formation in an Amine-Functioned Mesoporous Silica Surface
Aldol condensation is an important and typical class of carbon−carbon bond forming reactions in which amines can be used as catalysts. The reaction is also a common target in heterogeneous organocatalysis and has been the focus for the development and improvement of selective and reusable solid catalysts. In this study, Iowa State University and the Instituto de Química (Brazil) investigated the aldol reaction catalyzed by an amine-substituted mesoporous silica nanoparticle (amine-MSN) surface using a large molecular cluster model (Si392O958C6NH361) combined with the surface integrated molecular orbital/molecular mechanics (SIMOMM) and fragment molecular orbital (FMO) methods. The study analyzed three different pathways for the carbinolamine formation. It included a computational investigation of a reaction process encompassing surfaces like the Mesoporous silica nanoparticles (MSN) material which have high surface areas, narrow pore size distributions, and easy functionalization with specific organic and inorganic groups. The group determined that the investigation should involve a reliable surface model that is able to take into account the effects on the uppermost layer of atoms as well as the subsurface.
The computational study found that the most likely mechanism involves the silanol groups actively participating in the reaction, forming and breaking covalent bonds in the carbinolamine step. The active participation of MSN silanol groups in the reaction mechanism leads to a significant reduction in the overall energy barrier for the carbinolamine formation. In addition, a comparison between the findings using a minimal cluster model and the Si392O958C6NH361 cluster suggests that the use of larger models is important when heterogeneous catalysis problems are the target.
“In order to model catalysis on solid surfaces, the use of larger molecular models is very important, as is the inclusion of bulk effects and the surrounding environment. To perform such complex calculations requires the use of novel computational approaches, such as those based on hybrid quantum mechanics/molecular mechanics (QM/MM) models or FMO approach. The FMO method is particularly appealing since the code is highly scalable and is therefore able to make efficient use of massively parallel computer systems,” according to Gordon Iowa State University.
For more detailed information on this study, see Batista, Zahariev, Slowing, Braga, Ornellas, and Gordon. “Silanol-Assisted Carbinolamine Formation in an Amine-Functionalized Mesoporous Silica Surface: Theoretical Investigation by Fragmentation Methods.” Journal of Physical Chemistry, 15 December 2015.
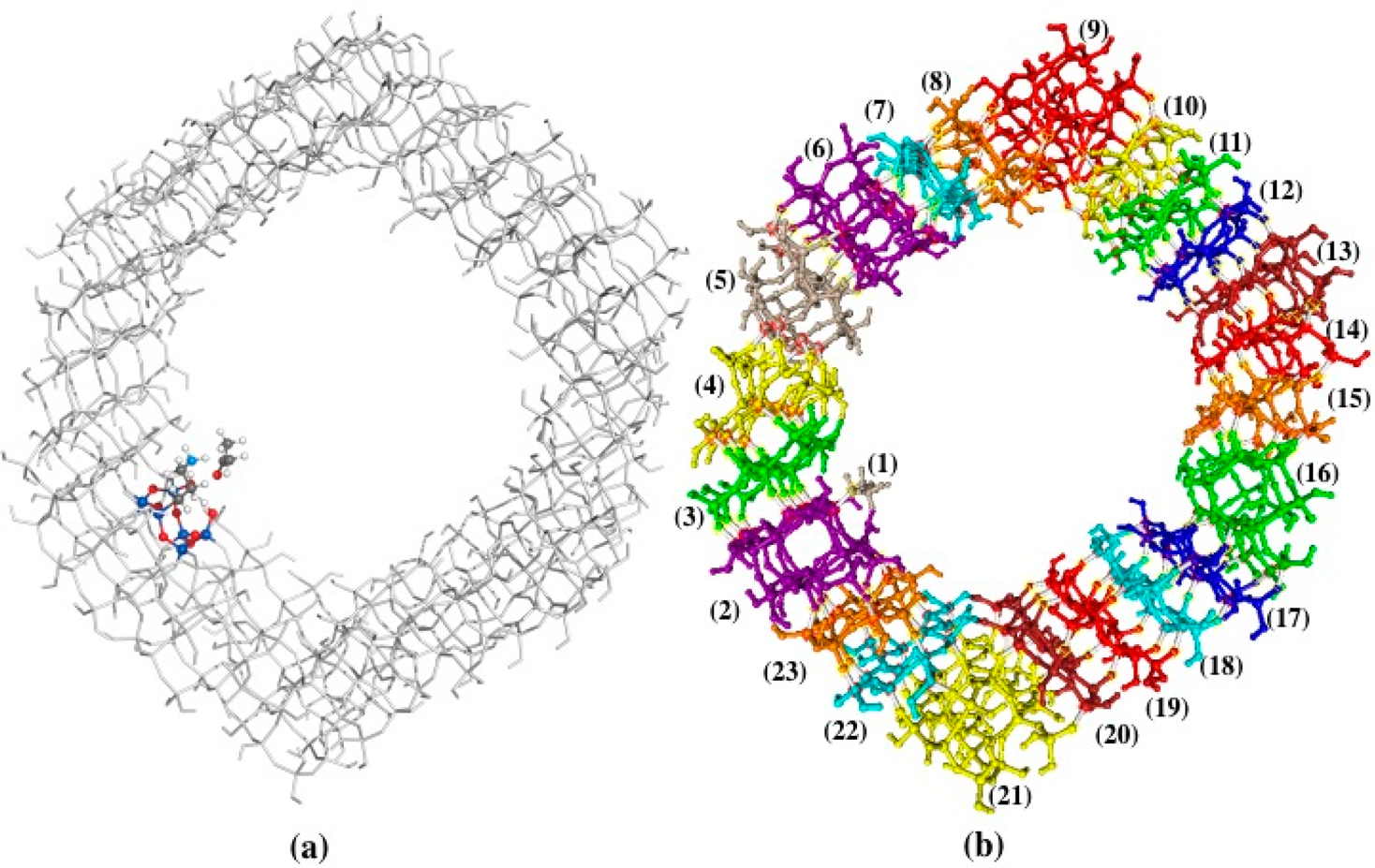
Figure 2. MSN surface model. (a) QM/MM molecular cluster for the primary-amine-MSN surface in the SIMOMM calculation. The QM atoms are colored: O atoms are red, Si atoms are dark blue, H atoms are white, and N atom is light blue. (b) Fragmentation scheme in the FMO calculation of the amine-substituted MSN surface; each fragment is indicated by a different color and number.
Introducing the Moscow State HPC System
Moscow State uses RSC PetaStream hardware installed in the Joint Supercomputer Center of Russian Academy of Sciences (JSCC) which are early adopters of the Intel Xeon Phi technology – the first system with Xeon Phi was installed in 2012. [3] RSC PetaStream is an ultra-high density HPC solution based on Intel Many Integrated Core (MIC) architecture. Each RSC PetaStream compute module is actually a server equipped with eight Intel Xeon Phi chips. In this solution, the CPU is used mainly for service purposes and is specially designed for a high MIC-to-MIC connection rate with the help of an FDR Infiniband network. As a result, the RSC PetaStream solution provides an almost uniform compute grid of up to 1024 Intel Xeon Phi modules per rack. In the Intel Xeon Phi coprocessor 7120D, it results in 1.2 PFLOPS/rack peak performance. Please refer to RSC PetaStream Ref. [4]. See technical details about RSC PetaStream hardware in Ref. [5].
How Moscow State is Optimizing GAMESS Code
The goal of Moscow State University is to revise and rewrite GAMESS code to improve the performance and scalability on the modern parallel architectures such as Intel Xeon Phi coprocessors. Their work will focus mostly on Hartree-Fock (HF) and density functional theory (DFT) methods. Next, parallelization strategies of FMO methods will be studied to maximize performance on this method on the Intel Xeon Phi coprocessor.
In GAMESS, the HF and DFT algorithms are parallelized using MPI and sockets. The current implementation imposes a high memory footprint when trying to use it on many core architectures with a small amount of onboard RAM such as an Intel Xeon Phi coprocessor. Mironov indicated, “In our current study, we developed hybrid OpenMP/MPI versions of the HF and DFT algorithms to overcome this limitation. We have studied different variants of workload distribution to achieve good load balance both between threads and between MPI tasks.
“Also, we focused on the optimization of serial performance. Our team tuned the code for better cache and memory usage as well as for vectorization. Our results show between a 30 percent to 150 percent speedup (depending on the workload specifics) for code optimized to run on an Intel Xeon Phi coprocessor versus the original code of the for the HF energy calculation. Code scaling is increased as well (20 percent to 100 percent improvement) that allows us to utilize more of Intel Xeon Phi compute power.” The following figure shows results of speedup.
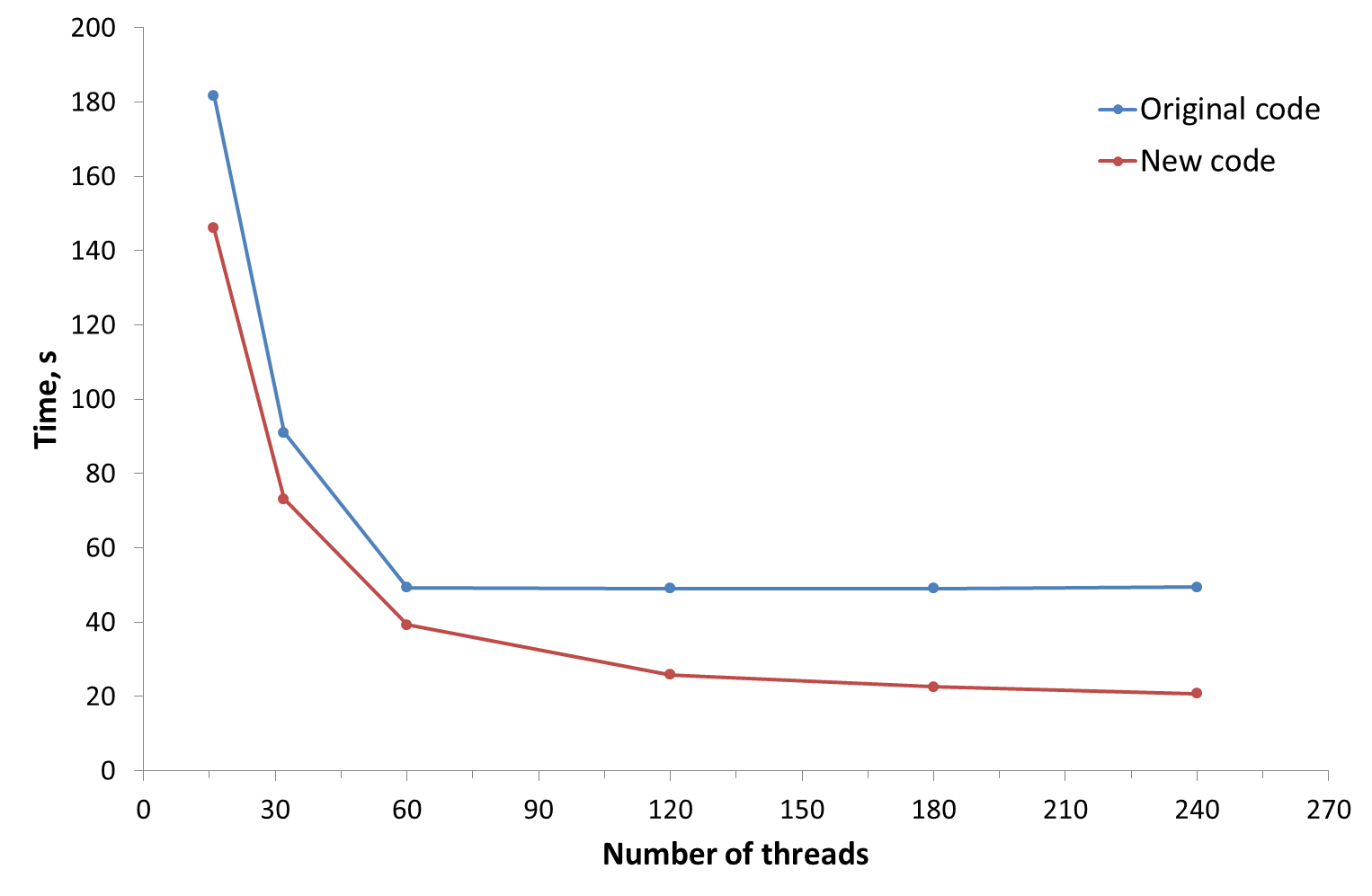
Figure 2. Strong scaling of single HF step performance for the Li6 cluster (cc-pVQZ basis set) on an Intel Xeon Phi coprocessor 7120D. Courtesy of Dr. Vladimir Mironov, Moscow State University.
HPC Software Tools Used in Optimizing Quantum Chemistry Simulations
Both Iowa State and Moscow State use a variety of tools such as Intel compilers and Intel VTune to identify vectorization targets in performance-critical sections of GAMESS code or improve vectorization. They use OpenMP because it allows them to do threading. Iowa State indicates that performance is assessed against existing codes and performance bottlenecks are determined using tools such as the TAU Tuning and Analysis Utilities Package which profiles parallel, threaded programs and maintains performance data for each thread, context, and node in use by an application. Both groups report using OpenMP and MPI so any optimization done for cache utilization, memory, or vectorization will benefit both the Intel Xeon processor and Intel Xeon Phi coprocessor. The same optimization approach will benefit the future Intel Xeon Phi processor code name Knights Landing.
How HPC Will Aid Molecular Dynamics Research in the Future
There is a process in place for submitting new GAMESS US code for review. Contributions to GAMESS go through a very extensive vetting process using the git/gerrit system. Once a submission passes, it is merged into the main branch and ultimately released. Work that the Iowa State and Moscow State teams are doing to optimize GAMESS code to run efficiently on Intel Xeon Phi coprocessors will be available to all users once it is reviewed and approved.
Gordon said, “There is a push in the U.S. driven by research by the Department of Energy and the National Science Foundation to move toward exascale computing. This cannot happen unless research finds a way to significantly reduce energy and the power footprint used in large computations. If we don’t find a way to do that, then the cost of operating these large supercomputer computations will be too expensive.
In our existing work at Iowa State, we have already seen that the Intel Xeon Phi coprocessor speeds up the calculations and uses low power. Our group at Iowa State purchased some Intel Xeon Phi 7120P coprocessors which allow you to modify the frequency. We will be doing research on how much you can reduce the clock speed without harming the performance time-to-solution. Our goal is to reduce power usage so that researchers can do more complex parallel computing calculations and simulations in an efficient manner, without increasing the power footprint.”
Mironov said, “Theoretical and quantum chemistry are traditional heavy users of compute resources available. As more research is done in the biochemistry area, we see a dramatic increase in compute power demand. Modern quantum chemistry is being developed in the area of large molecule simulation, with notable examples of QM/MM and FMO approaches. Currently, it is feasible to simulate properties like spectra of large protein molecule or reaction rate constant for an enzyme. We hope that in the future it will be more feasible to address problems involving protein complexes (like membranes or even viruses) or enzymatic reaction constants for large portions of metabolic network. For smaller systems, supercomputers allow researchers to characterize their properties even better by gathering vast amount of statistical data (e.g., using ab initio molecular dynamics).
“For the foreseeable future, supercomputers and large compute clusters will be necessary to conduct state-of-the art theoretical research in quantum chemistry simulations, especially for molecules like proteins,” Mironov continued. “Intel is doing a great deal of work in terms of serving the high performance computing ecosystem, including processors and manycore processors (Intel Xeon Phi coprocessor, Intel Xeon Phi processor code name Knights Landing), compilers, debuggers and profilers, MPI, storage, networks.”
About the author
Linda Barney is the founder and owner of Barney and Associates, a technical/marketing writing, training and web design firm in Beaverton, OR.
Read more:
Modified NWChem Code Utilizes Supercomputer Parallelization
Speeding up Molecular Dynamics: Modified GROMACS Code Improves Optimization, Parallelization
Optimized, Parallelized Software Enhances Quantum Chemistry Research
DL_POLY Molecular Dynamics Code Changes Improve Optimization and Parallelization
Supercomputers Aid in Quantum Materials Research
Molecular Dynamics Research Enhanced by More Scalable LAMMPS HPC Code
References:
Iowa State
https://software.intel.com/en-us/articles
http://www.msg.chem.iastate.edu/
http://www.msg.ameslab.gov/GAMESS/capabilities.html
http://www.msg.ameslab.gov/gamess/gamess_manual/gamess-05.pdf
http://www.sciencedirect.com/science/article/pii/B9780444517197500846
http://onlinelibrary.wiley.com/doi/10.1002/jcc.540141112/full
Moscow State
2. http://www.msg.ameslab.gov/gamess/
3. www.jscc.ru
5. Andrey Semin , Egor Druzhinin, Vladimir Mironov, Alexey Shmelev, Alexander Moskovsky “ The Performance Characterization of the RSC PetaStream Module” Volume 8488 of the series Lecture Notes in Computer Science pp 420-429
6. Vladimir Mironov, Maria Khrenova, Alexander Moskovsky“ On Quantum Chemistry Code Adaptation for RSC PetaStream Architecture” High Performance Computing Volume 9137 of the series Lecture Notes in Computer Science pp 113-121


