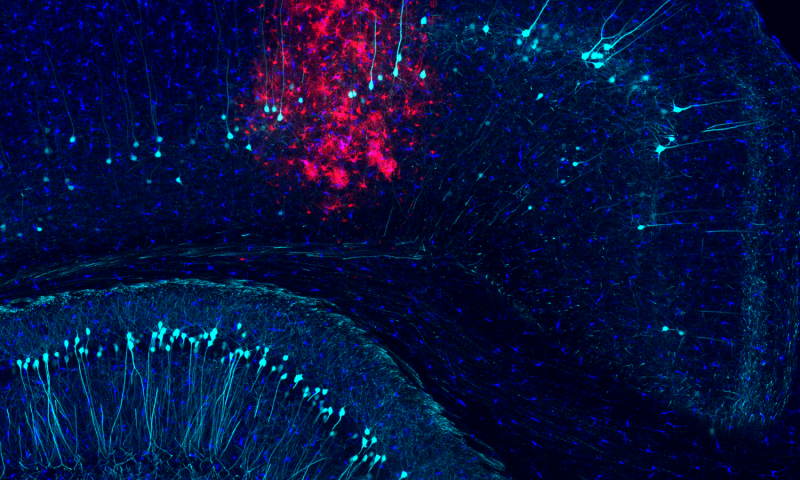
Salk Institute researchers discovered that inhibiting a protein called phospholipid scramblase 1 (PLSCR1) controls the infected cell’s antiviral response and provides long-term protection from immune attack and excessive inflammation. Salk Institute researchers discovered that inhibiting the protein PLSCR1 controls the infected cell’s antiviral response and provides long-term protection from immune attack and excessive inflammation. In the cortex of a mouse, adenovirus-transduced cells (red) appear among labeled microglia (dark blue) and subsets of neurons (light blue). (Credit: Salk Institute)
Normally when we think of viruses, from the common cold to HIV, we want to boost people’s immunity to fight them. But for scientists who develop therapeutic viruses (to, for example, target cancer cells or correct gene deficiencies) a more important question is: How do we keep people’s natural immune responses at bay? In these cases, an overenthusiastic immune response actually undermines the therapy.
Salk Institute researchers discovered that inhibiting a protein called phospholipid scramblase 1 (PLSCR1) controls the infected cell’s antiviral response and provides long-term protection from immune attack and excessive inflammation. The results, described in the January 19, 2017, issue of Neuron, hold promise both for virally delivered treatments and inflammatory conditions like infections; autoimmune disorders such as lupus; or neurodegenerative diseases such as Alzheimer’s disease.
“Normally, the immune system will quickly recognize and act upon potential threats such as virally infected cells,” says Axel Nimmerjahn, assistant professor in Salk’s Waitt Advanced Biophotonics Center and senior author of the new paper. “But in targeting PLSCR1, we’ve effectively shielded infected cells from immune attack and increased gene expression from an engineered virus from a few days or weeks to at least six months, creating the potential for much longer-lasting therapies.”
Because viruses have evolved such effective methods for finding their target and utilizing the host cells’ own molecular machinery, scientists are modifying viruses into long-lasting therapeutic vehicles that can deliver drugs to specific cells or tissues or selectively target and destroy tumors. But, since our immune systems don’t discriminate between destructive and therapeutic viruses, researchers must devise ways to keep the immune system from destroying the helpful versions.
To better understand the brain’s immune response, the Salk team injected an engineered adenovirus (often used to deliver gene therapy) into mouse brains and monitored both the infected and immune cells‘ signaling and protein production in response to infection. Infected cells put out a variety of distress calls, including a molecular flag called phosphatidylserine that summons microglia, specialized immune cells in the brain. Microglia assess the situation and then decide whether to leave the infected cells alone or destroy them. Leaving the distressed cells alone risks having them cause collateral damage to other cells, while destroying them eliminates potentially critical brain cells, many of which don’t regenerate. To better understand the calculus of this decision, the Salk team manipulated levels of the various proteins involved in the cell’s intra- and extracellular communication, including PLSCR1.
Surprisingly, ramping down PLSCR1 caused a wide variety of immune-related changes. Besides keeping microglia at bay, less PLSCR1 resulted in reduced production of inflammatory molecules called cytokines, whose typical role is to recruit more immune cells to the fight.
“When we saw how much inhibiting PLSCR1 reduced the inflammatory response, we immediately wondered if this mechanism could apply more broadly, not just to virus infection of the brain, but to other types of infections or even autoimmune diseases,” says Yusuf Tufail, a former Salk postdoctoral fellow and first author of the paper.
The protective effects of inhibiting PLSCR1 lasted for all time points investigated (up to six months); none of the other manipulated proteins had such a pronounced result. Because PLSCR1 is present throughout the body, not just in the brain, researchers plan to investigate the role of PLSCR1 in other inflammatory conditions.
“Given how complex the immune response is, and how many genes are regulated up or down in response to infection, it was amazing to find a single protein that controls so many pathways,” says Nimmerjahn. “Imagine a small molecule inhibitor that a patient could take to curb excessive inflammation. This could have a hugely beneficial effect on many disease outcomes.”

Yusuf Tufail (front) and Axel Nimmerjahn. (Credit: Salk Institute)


