Researchers have made significant strides in predicting a protein’s structure from its sequence using large language models. However, this method hasn’t been as effective for antibodies, primarily due to their hypervariability. This makes it challenging to identify treatments for SARS-CoV-2 and other infectious diseases.

MIT researchers have developed a computational technique that more accurately predicts antibody structures, especially in their hypervariable regions, which are essential for binding to pathogens. While this marks an advancement in protein modeling, the team highlights that obstacles remain before all antibody-folding puzzles can be resolved. Nevertheless, the new method may assist drug developers in identifying promising vaccine and therapeutic candidates early in the research process. “Our method allows us to scale, whereas others do not, to the point where we can actually find a few needles in the haystack,” says Bonnie Berger, the Simons Professor of Mathematics and head of the Computation and Biology group in MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL). “If we could help to stop drug companies from going into clinical trials with the wrong thing, it would really save a lot of money.”
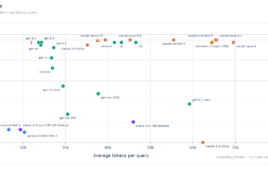
The model, AbMap (Antibody Mutagenesis-Augmented Processing), adapts large language models to focus on the highly variable tips of antibodies. Traditional AI tools, such as AlphaFold, struggle in these regions because of the sheer diversity of antibody sequences. By combining data from thousands of known antibody structures with binding-strength measurements for specific antigens, the researchers train AbMap to predict structures and assess how strongly an antibody might bind its target.
In one demonstration, the team generated millions of antibody variants for the SARS-CoV-2 spike protein and used AbMap to narrow them down to a handful of likely strong binders. Follow-up experiments with collaborator Sanofi showed that 82% of these picks offered better binding strength than the original antibody. Rohit Singh, a former CSAIL research scientist who is now an assistant professor of biostatistics and bioinformatics and cell biology at Duke University, and co-lead author, notes, “They don’t want to put all their eggs in one basket. They would rather have a set of good possibilities and move all of them through, so that they have some choices if one goes wrong.”
The researchers also see the potential for analyzing entire antibody repertoires to understand why certain individuals — such as “super-responders” to HIV — mount effective immune responses. While the method makes structure prediction more efficient, the team acknowledges that, given the vast range of possible variants, it does not resolve all obstacles in antibody design.
“This is where a language model fits in very beautifully because it has the scalability of sequence-based analysis, but it approaches the accuracy of structure-based analysis,” Singh says.
The study appears in the Proceedings of the National Academy of Sciences and was supported by Sanofi and the Abdul Latif Jameel Clinic for Machine Learning in Health. Researchers from the Ragon Institute of MGH, MIT, Harvard, and ETH Zurich contributed.