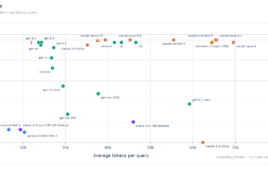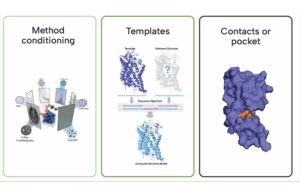
A Boltz-2 diagram as shown on https://www.rxrx.ai/boltz-2
A team from MIT CSAIL, the Jameel Clinic, and Recursion has released Boltz-2, an open-source Python-based biomolecular model that performs physics-level protein-ligand affinity predictions in approximately 18 seconds on a single consumer GPU. This task previously required hours or days on cluster hardware, often costing upwards of $100 per molecule. By releasing the model, weights, and full training pipeline under a permissive MIT license, the team provides unrestricted access to supercomputer-class modeling for academic and commercial R&D.
“The cost and time of precise affinity measurements is typically the limiting factor for the number of molecules and proteins we can test,” explained Gabriele Corso, MIT researcher and co-creator of Boltz-2. “Existing computational methods are unfortunately either too slow—this is the case for FEP, which is used in the field but can cost hundreds of dollars per prediction and take several hours to a day to run—or they are fast but far too inaccurate to be relied upon.”
At its core, Boltz-2 introduces several new developments:
- Joint structure-and-function: While previous models stop at predicting a molecule’s 3D shape, Boltz-2 jointly predicts the structure, its binding affinity, and B-factors (a measure of flexibility) in a single pass.
- Physical steering: The model uses a Feynman-Kac–style potential to guide its predictions away from physically impossible states like overlapping atoms or incorrect bond lengths. “Boltz-2 is the only model of its sort that produces quality structures that are physically plausible in nearly 100% of the cases,” stated MIT researcher Jeremy Volfand.
- Broader training data: Beyond the standard PDB, the model was trained on thousands of molecular dynamics trajectories and roughly five million public binding affinity assays, improving its ability to generalize.
Image drawn from the Boltz GitHub page
The model’s performance was validated across multiple industry-standard benchmarks. Those included hit-to-lead (FEP+ Benchmark) where it achieves a Pearson correlation of 0.62, approaching the accuracy of full physics simulations (0.72) but running approximately 1,000 times faster. Boltz-2 also also retrieved nearly twice as many true binders among the top-ranked molecules as the next-best baseline method on the Hit Discovery (MF-PCBA) benchmark. In addition, on the Real-World Generalization (CASP16) benchmark, Boltz-2 achieved an out-of-the-box correlation of 0.65 on unseen internal targets from Roche, outperforming other academic competitors.
“Binding affinity is core to developing a therapeutic, start to finish,” said Najat Khan, Chief R&D Officer at Recursion. “You want to bind to the right areas and you don’t want to bind to the wrong proteins to limit off-target effects, especially for small molecule drug discovery.”
The project’s open-source availability is core to its mission. Its predecessor, Boltz-1, grew a Slack community of over 1,300 users and saw 40 external code contributions in six months. This avid community is already pushing the tool to its limits, asking how to scale up virtual screenings, model complex antibody interactions, and fine-tune predictions with proprietary data. “The MIT license basically says you can take the code and do whatever you want with it,” noted co-creator Gabriele Corso in the press conference. “99.9% of drug developers and biologists are outside of companies like Isomorphic [Ed note: Isomorphic Labs is Alphabet’s drug discovery spin-off from Google DeepMind]. Part of the reason we are releasing this fully open source is because we want all of these biologists to have access to it.”
For years, the modest accuracy of AI affinity predictors has kept them on the sidelines of many drug discovery campaigns. A Pearson correlation of 0.62, however, approaches a threshold where computational predictions can become genuinely useful for hit-to-lead optimization. Yet the team acknowledges that a 0.62 correlation does not eliminate the need for wet-lab validation. Peer review for the work is pending, and the model’s accuracy is less reliable for protein families with sparse public data.