Environmental system designers decide where to place sample points for particle monitoring. However, the process is not always straightforward. There are several guidance documents that offer advice on what processes need to be monitored, along with advice on suitable distances from the process being monitored.
Particle counting in pharmaceutical applications is clearly segregated into one of three categories, each requiring a different approach:
Certification is measuring a cleanroom for a standard. The only standard recognized worldwide is ISO14644-1, “Classification of air cleanliness by particle concentration,” which defines how a cleanroom performs and its ability to show uniformity across the entire space. This is done irrespective of the activities performed in it. The specifics of the assessment may vary slightly for FDA or EU GMP regulations, but the underlying methodology is standard.
Qualification is the process of analyzing risk assessment for the activities in the room. Qualification follows grid methodology testing methods. Particle counts are measured in both operational and at-rest states; however, the operational data is the most valid.
Monitoring is the ongoing sampling of the cleanroom on a frequency relative to the degree of control required to prove management over risk to the finished product. The number of sample points and their location is determined by risk assessment, and the qualification and certification process.
Certification
Certification demonstrates that the entire area meets a specific ISO classification by particle concentration. Irrespective of the final use of the room, the design and implementation of the filtration system are considered. The international standard indicates that a cleanroom tested to meet compliance for ISO 5 standards meets that standard independent of geography and regulatory aspects (i.e. FDA or EU GMP).
There are many resources to prove ISO compliance. Using the example of a classic filling machine (Grade A/ISO 5) within a Grade B (ISO 7) background area, basic testing rules can be demonstrated.
1. The number of sample points is based on the area.
a. Calculate the area of Grade A/ISO 5.
b. Calculate the area of Grade B/ISO 7.
2. Sample point placement for Grade A (ISO 5) area:
a. The sample points must all be equidistant and at work height, irrespective of the activity at the location of their placement.
b. Samples are taken in a grid pattern at the identified locations. Derive the minimum number of sampling locations, NL, from ISO 14644-1 Table A.1, which provides the number of sampling locations related to the area of each cleanroom or clean zone to be classified, and provides at least 95 percent confidence that at least 90 percent of the cleanroom or clean zone area does not exceed the class limits.
c. PASS/FAIL criteria are calculated for ISO and EC GMP Annex 1. It is recommended to have both sets of data, as the FDA requires ISO14644-1, and the EC requires Annex 1 data points (although the EC data would suffice for the FDA).
3. Sample point placement for Grade B (ISO 7) area:
a. Repeat the steps used for Grade A (ISO 5) area.
b. It may be more difficult to determine the locations of the sample points due to the room’s unusual shape. Derive the minimum number of sampling locations, NL, from ISO 14644-1 Table A.1. This table provides the number of sampling locations related to the area of each cleanroom or clean zone to be classified, and provides at least 95 percent confidence that at least 90 percent of the cleanroom or clean zone area does not exceed the class limits.
4. Final report marks end of certification phase.
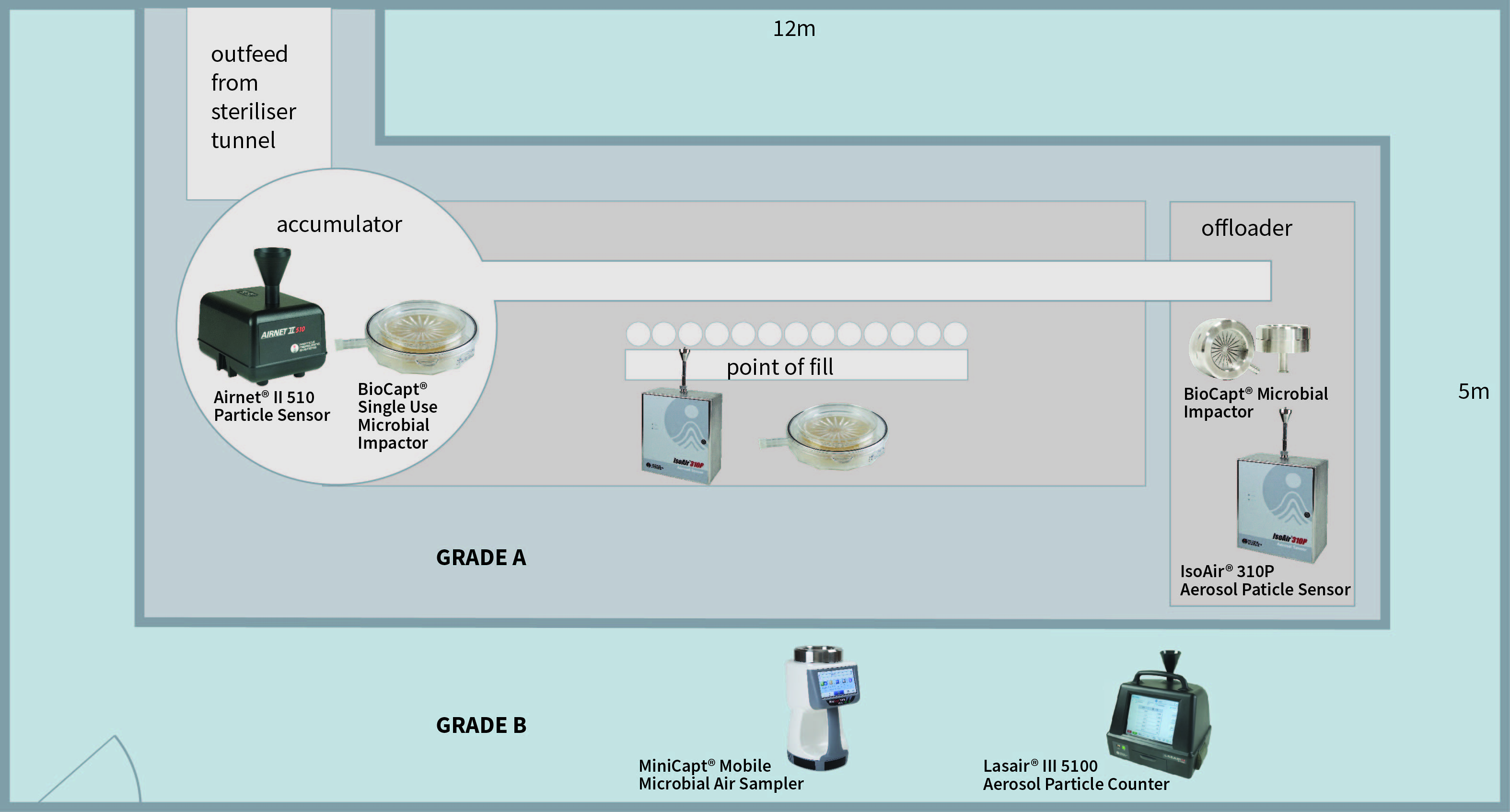
Impactors and sensors should comply with ISO 14698-1, certification standards set for biocontamination control.
Qualification
The qualification phase considers the risks to the quality of the finished product. Each activity must be considered and measured. Continuing with the example of the filling line, let us consider the accumulator table at the exit of the sterilizer tunnel. The risk is that glassware (vials/bottles) is exposed to the open environment and operator. Therefore, it is possible for contamination to fall into clean vials/bottles prior to filling. Operator intervention and the shifting of glassware cause turbulent air movement on the table, impacting contamination risk. Therefore, the following actions should be taken:
1. Divide the area of risk into a 3×3 or a 4×4 grid. If activity can occur at several levels, then each level (working height, +150 mm from work height, and +300 mm from work height) must be considered.
2. Take a particle sample at the center of each of the grid squares and on each level.
a. Samples are taken during ‘At Rest’ and ‘Operational’ states. It may be required to work around an activity or operator to gain suitable data. Slight movement of sample points within the grid square is acceptable. A location is invalid if found to impede normal activities.
3. Together, all collected samples provide a particle map of the pharmaceutical activity.
Each of the key functions within the cleanroom (filling point, stoppering, general background activities, etc.) should be analyzed accordingly.
Monitoring
The location of monitoring points should be based upon a formal risk assessment (FMEA, FMECA, or other risk analysis tools) using data from the certification and qualification testing. Other factors (equipment interference, mounting points, operator impedance and intervention) contribute to selecting the final location for the sample probe.
During a post-analysis assessment of the sample point, it may be determined that a location is directly in line with where the operator needs to make routine system adjustments, thus impeding operator activities. In such a case, the sample point must be moved, and the second-highest ranking sample point should be selected.
The isokinetic sample probe should face into the air stream and the minimum length of tubing should be used. Although different manufacturers claim specific lengths of tubing can be used with their particle counter, this is typically a function of vacuum pump dynamics, and not of particle transportation. Particles of 0.5 μm move freely in long lengths of tubing. However, 5.0 μm particles do not have this same mobility. As 5.0 μm particles are a greater concern, the tubing should be maintained at its shortest recommended lengths1. Particle Measuring Systems quotes maximum tubing lengths based upon the same conditions of airflow, and has a recommended maximum length of 3m. However, for pharmaceutical particle systems we advise a reduced recommended length of 2m to ensure transportation of the larger particles.
From the FDA’s Aseptic Processing CGMP Guideline: “Air in the immediate proximity of exposed sterilized containers/closures and filling/closing operations would be of appropriate particle quality when it has a per-cubic-meter particle count of no more than 3520 in a size range of 0.5 μm and larger when counted at representative locations normally not more than 1 foot away from the work site, within the airflow, and during filling/closing operations. This level of air cleanliness is also known as Class 100 (ISO 5).”
The frequency of sampling should reflect the risks and follow from the FDA guidelines on sterile manufacturing and the EC-GMP Annex 1 Guide. Particle monitoring should be automated and maintained in a continuous state when glassware and products are exposed.
References
1. http://www.pmeasuring.com/en/knowledge-center/application-notes/particle-loss-in-transport-tubing
Gilberto Dalmaso, PhD, is Global Aseptic Process Development Manager with Particle Measuring Systems. Gilberto has over 25 years’ experience in pharmaceutical microbiology and sterility assurance, and currently focuses on pharmaceutical microbiology and aseptic processes, microbiological contamination control and rapid microbiological methods, Quality by Design, and PAT in microbiology. [email protected]
Mark Hallworth is Pharmaceutical Sales Manager with Particle Measuring Systems. Mark has managed the design, installation, and validation of more than 300 environmental monitoring system projects worldwide. He has lectured for the PDA, ISPE, and other pharmaceutical societies on environmental monitoring and GMP compliance design and validation. [email protected]; www.pmeasuring.com



