
There is no limit to the need for massive compute power to drive simulations of molecular and atomic systems. The drive is ongoing to improve parallelization for higher compute performance that lets research models operate on more particles or longer timescales. As a result, more sophisticated simulations become possible, accelerating research and improving the quality of results.
Scientific computing problems such as molecular-dynamics simulations require coordinated engineering of the system hardware and the software that runs on it. To that end, Sandia National Laboratories works closely with industry on the development of the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS), in addition to acting as custodian for the open-source code.
One important product of that collaboration has been to optimize LAMMPS for standards-based many-core processors. As an alternative to special purpose approaches, developers benefit from a more open approach that does not tie them to a single compiler or a single architecture. Instead, their code can be optimized for a variety of hardware, ranging from many-core processors to the larger cores on general-purpose CPUs.
Broad-Based Collaboration to Advance LAMMPS
A long-running collaborative relationship between Sandia Labs and Intel benefits research organizations that utilize LAMMPS code. A key example of that collaboration lies in the development of Intel’s compiler and math library, with Sandia typically becoming involved in each new version of the tools in their earliest days. Working on emulated next-generation hardware, Sandia runs millions of lines of code through alpha-stage compilers, helping detect bugs as well as enhancing LAMMPS code to take best advantage of upcoming compiler versions.
Likewise, Intel makes significant contributions to the LAMMPS project, such as enhancing the core kernels that perform math operations to ensure that they vectorize cleanly. Intel’s Mike Brown has done extensive work to enable LAMMPS for vectorization. Vectorization can result in significant gains for newer processors. For example, with Intel® Xeon Phi™ x200 series processors and future Intel® Xeon® processors, Intel® AVX-512 instructions dramatically improve performance, allowing a register to hold twice as many data elements as previous generations of Intel AVX: eight double-precision or sixteen single-precision floating-point numbers.
Intel AVX-512 also contains new capabilities that can improve vectorization of molecular-dynamics algorithms and others. To better exploit newer Intel® processors, the LAMMPS team has implemented approaches that use conflict-detection instructions (Intel AVX-512CD), which are supported in the Intel Phi processor formerly code-named Knights Landing and future Intel processors.
Loops with data dependencies can create issues when a vector containing calculation results needs to be written back into memory: it is possible for multiple values to conflict with each other, needing to write to the same location. To avoid performance impacts associated with these conflicts while still keeping as much code as possible running with vectorization, Intel AVX-512CD provides instructions that detect data conflicts when working with 512-bit vectors.
Importantly, a single version of the LAMMPS software package is portable across both Intel Xeon Phi processors (with up to 72 cores per socket) and general-purpose Intel Xeon processors.
Performance Improvements on Multiple Hardware Architectures
As optimizations of LAMMPS code using Intel AVX-512 continue through the collaboration between Intel and Sandia, Intel has performed initial performance testing to gauge the effectiveness of these efforts on various types of hardware. To provide a broad comparison, the testing included both the standard LAMMPS code and the optimized version, running on both previous-generation and current-generation versions of the Intel Xeon processor, as well as the current-generation Intel Phi processor.
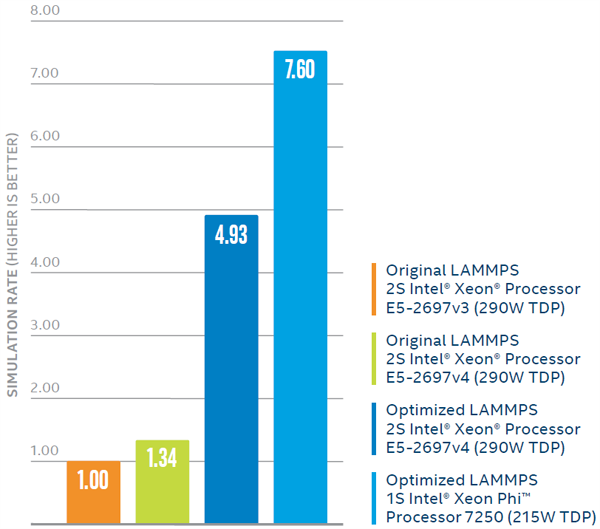
Improvements in LAMMPS performance from software optimizations and hardware advances.1
Comparing performance of the standard LAMMPS code on the previous-generation Intel Xeon processor versus the current-generation one showed a speedup in simulation rate of 1.34x. While significant, that performance benefit is far less than the speedup achieved using the code specifically optimized for Intel® architecture. Using that optimized code on the same Intel Xeon processor E5-2697 v4 produced a speedup of approximately 3.68x, compared to standard LAMMPS.
Performance on the Intel Xeon Phi processor is higher still, with an additional more than 1.5x speedup compared to the identical code running on the current-generation Intel Xeon processor. The fact that this testing was performed using a single code base on both Intel Xeon and Intel Xeon Phi processors represents a significant step forward. By enabling programming for many-core processors using the same programming methods as general-purpose processors, development is significantly simplified for far greater code flexibility.
The advantages are clear in terms of running simulations faster using code optimized as part of the collaboration between Intel and Sandia. In addition to the performance and flexibility benefits, those optimizations also enhance the efficiency of the code, as measured in performance per watt. For details, see the Intel technology brief, “Boost Performance and Energy Efficiency on HPC Workloads with Intel® Xeon Phi™ Processors.”
Tangible Benefits for Research
Outside the test lab, these advances promise better results on molecular-dynamics simulations used in a variety of disciplines. To help safeguard the integrity of those research efforts, Sandia and Intel rigorously test the newly optimized LAMMPS code at each stage to ensure that particular inputs produce the correct outputs. This testing helps give the scientific computing community confidence that it can rely on the improved code to produce high-quality results, in addition to increased performance and energy efficiency.
Optimization of LAMMPS code provides significant value to many types of research, including those described briefly below.
Case Study: Climate Research
Accelerated simulations using LAMMPS benefit climatic research underway at the US National Science Foundation’s Center for Aerosol Impacts on Climate and the Environment (CAICE). Researcher Yuqing Qiu works on the use of computer simulations to investigate the interplay between microscopic structure, dynamics and phase transformations in aerosols, with the goal of accurately predicting the impact of aerosols on climate and our environment.
Her research is supported by the collaborative work between Intel and Sandia to enable greater sophistication in LAMMPS simulations. Qiu reports that, “The Intel package increases the speed of our simulations and makes a huge difference to the kinds of problems we can work on. It enables us to simulate much longer time scales and larger systems, and bring the real-world chemical complexity into simulations.”
Case Study: Pharmaceutical Development
Accelerated simulations throughout a hypothetical drug-development process help show the potential value of ongoing optimization of LAMMPS for emerging generations of processors. An early-stage requirement in designing a new therapeutic compound is to identify potential targets for the drug. For example, research might focus on identifying proteins within a pathogen that are vital to replication, while also being dissimilar enough from proteins the human host depends on. Disrupting the function of that protein could hold promise for action by a potential new drug.
Having identified the targets of interest, simulations can study interactions between potential therapeutic compounds and those proteins. Being able to carry out those studies virtually instead of with actual pathogens and compounds of interest has obvious value in terms of time and cost requirements. In fact, it also allows studies to be carried out using compounds that don’t exist in the physical world. By enabling such virtual studies to proceed more quickly, with larger data sets and longer timescales, Sandia and Intel hope to give pharmaceutical researchers the tools to respond more rapidly to novel pathogens, create more robust therapeutics for existing afflictions, and improve quality of life.
Conclusion
Optimization by Sandia and Intel of the LAMMPS software package for molecular-dynamics simulation is a strong example of the potential from collaboration between the public and private sectors. By embracing an open-systems programming approach, LAMMPS is helping set the stage for programming that crosses boundaries between systems based on many lightweight cores, as well as those with smaller numbers of more powerful cores.
Optimizations of LAMMPS for Intel-based hardware show the viability of a flexible development approach across architectures. As future processors and instruction set architectures are introduced, parity between many-core and multicore programming will continue to yield dividends, measured in both flexibility and improved research outcomes.
Matt Gillespie is a technology writer based in Chicago. He can be found at www.linkedin.com/in/mgillespie1.
Source: https://software.intel.com/sites/default/files/managed/8b/96/LAMMPS_Brief.pdf.
Node and Software Configurations Measured by Intel: The benchmarks were run by Intel on two-socket Intel® Xeon® E5-2697 v3 (Haswell) and E5-2697 v4 (Broadwell) processors and one-socket Intel® Xeon Phi™ 7250 (Knights Landing) processors. The respective BIOS versions were SE5C610-86B-01-01-5008-081020151115, SE5C610-86B-01-01-0015-012820160943, and GVPRCRB1-86B-0009-D10-1602121844. The respective Red Hat Enterprise Linux* versions were 6.6, 6.7, and 6.7 with kernels 2.6.32, 3.10, and 3.10. The respective processor steppings were 2, 1, and 1. The respective DDR4 configurations were 8x8GB 2133MHz, 8x16GB 2400MHz, and 6x16GB 2400MHz. The cluster/snoop modes were “Home,” “Home,” and “Quadrant” with the Knights Landing node configured in “Flat” mode so that MCDRAM was not available as an L3 cache. The binaries were built using the “intel cpu” and “knl” makefiles provided with LAMMPS. All optimization flags were the same aside from the target ISA. The “22 Mar 16” version of LAMMPS was compiled with Intel® 16.0.2 compilers using version 5.1.2.150 of Intel MPI.



