Next-generation sequencing (NGS) tools have been a game-changer in the halls of drug discovery, where rapid and affordable sequencing has enabled faster characterization of samples, the ability to interrogate individual cells, and more frequent monitoring of cell lines for genetic drift, among other capabilities.
 While these platforms have contributed to significant advances in drug discovery and development, one limiting factor has been consistent across nearly all NGS technologies: their inability to generate a comprehensive view of the genome. These same platforms were applied to the human reference genome for years without being able to characterize the intractable 8% of the genome that was only recently revealed in the Telomere-to-Telomere (T2T) consortium assembly.1
While these platforms have contributed to significant advances in drug discovery and development, one limiting factor has been consistent across nearly all NGS technologies: their inability to generate a comprehensive view of the genome. These same platforms were applied to the human reference genome for years without being able to characterize the intractable 8% of the genome that was only recently revealed in the Telomere-to-Telomere (T2T) consortium assembly.1
Conventional NGS tools miss many genomic elements that are important for biopharmaceutical development. For example, short-read sequencers struggle to represent structural variants, repetitive DNA, maternal and paternal haplotypes, and other elements too long to capture in just 300 base pairs. In addition, short-read sequencers that rely on amplification typically cannot detect epigenetic marks, as methylation gets stripped away in the sample preparation process.
Recently, scientists have deployed newer sequencing tools with unique features that make it possible to capture these commonly missed elements. Long-read sequencing can overcome challenges associated with short-read sequencing, while native DNA analysis allows for detecting methylation status with a much faster turnaround time. Several published studies have demonstrated the utility of these alternative technologies for drug discovery and development applications.
More comprehensive genomic analysis will be useful for many aspects of biopharmaceutical development, but the need for an accurate and complete representation of DNA is more pressing than ever in the era of gene editing. As editing technologies are developed for more clinical applications, having a comprehensive view of relevant genomic elements will be critical for making these treatments as effective and successful as possible.
What you’re missing matters
The T2T reference genome released in 2022 finally finished the human genome assembly, recovering the last 8% of DNA sequence that had never been accessible. Among the previously missing regions were segmental duplications, repetitive elements, and other components that could be relevant for clinical diagnosis or therapeutic development.
Segmental duplications, for instance, have been associated with neurological disorders, making these hard-to-characterize elements important as potential biomarkers or drug targets. It’s a similar story for repeat expansions, which are linked to Huntington’s disease, fragile X syndrome, and other neurological conditions.
But complicated and less complicated elements, such as structural variants, have proven intractable with standard NGS platforms. Biomarker discovery was one of the earliest beneficiaries of NGS tools, so it has mostly been limited to single nucleotide variants (SNVs) because these single-base changes are the easiest to spot with short-read NGS technologies. Much of drug discovery analysis is focused on SNVs for this reason. But as more complex variants are detected and associated with a wide variety of diseases, scientists at pharmaceutical and biotech companies will be more productive if they can expand their biomarker discovery efforts beyond SNVs.
Methylation detection is another area ripe for a new approach. A few existing drugs target methylation, but epigenetic changes are linked to many more diseases and disorders. The field of epigenomics is rapidly growing, and the latest research findings could be helpful in drug discovery and development. Better technology platforms are needed to characterize methylation and its effects on gene expression accurately and cost-effectively.
Innovative approaches to DNA sequencing could also bear fruit in newer clinical research areas, such as liquid biopsy and single-cell studies. For these applications, sequencers must be highly accurate and sensitive — able to sense signals masked in a sample.
Recent advances
Novel sequencing technologies are being used by some scientists in drug discovery and development, and their results demonstrate the tremendous potential to advance clinical research.
In one study, scientists at DeCODE Genetics used a nanopore-based native long-read sequencing technology to perform whole genome sequencing of 3,622 people in Iceland.2 The project was designed to improve the detection of structural variants in human genomes and proved successful: scientists could identify more than 22,000 structural variants, on average, per person. They associated those variants with disease phenotypes and discovered a previously unknown link between lower LDL cholesterol levels and a rare deletion in the PCSK9 gene, resulting in an enticing new target for cholesterol-lowering drug development programs.
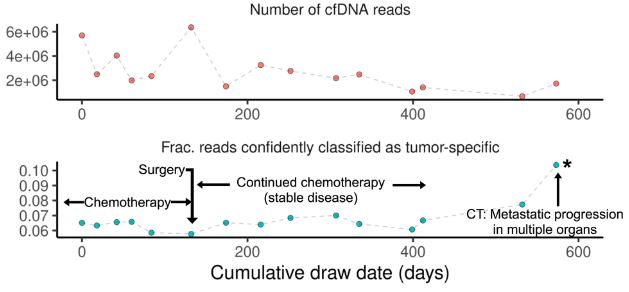
Another interesting study came from scientists at Stanford University who focused on methylation profiling of cell-free DNA. They chose nanopore sequencing for this project because it can detect methylation status directly as it sequences native DNA, with no bisulfite conversion or additional analysis step required.3 The approach involved single-molecule sequencing to profile the methylomes of cell-free DNA samples collected from patients with cancer. For one sample, the technique generating as many as 200 million reads scientists noted was “an order of magnitude improvement over existing nanopore sequencing methods.” As part of the project, the team built a single-molecule classifier to pinpoint whether sequence reads represented DNA from a tumor or immune cell. This allowed them to compare methylomes originating from each cell type and to perform longitudinal monitoring of methylation changes in these patients-derived samples during cancer treatment. Such an analysis could be used in drug discovery efforts focused on methylation biomarkers and drug development, where noninvasive sample collection can maximize data gathered in a clinical trial. In addition, this nanopore technology enables sequencing one sample at a time, enabling faster turnaround time without the constraint of having to batch dozens or hundreds of samples with legacy short-read technologies.
 At the Amsterdam Cancer Center, scientists took their approach to cell-free DNA analysis. They evaluated using nanopore sequencing for cell-free DNA collected in plasma and urine samples from patients with lung or bladder cancer, respectively, focusing on time to results as an indicator of potential clinical utility.4 The team quantified copy number aberrations in the samples and identified key patterns in the cell-free DNA recovered, confirming that fractions of cell-free DNA derived from the tumors in each sample matched what they expected based on orthogonal technologies. Interestingly, they could recover many long DNA fragments originating from the tumors. All of this was done rapidly, with DNA characterization and analysis performed within 24 hours of sample collection. Again, this suggests potential utility in a clinical trial setting, where time to results is often important for understanding unexpected reactions to a candidate therapy.
At the Amsterdam Cancer Center, scientists took their approach to cell-free DNA analysis. They evaluated using nanopore sequencing for cell-free DNA collected in plasma and urine samples from patients with lung or bladder cancer, respectively, focusing on time to results as an indicator of potential clinical utility.4 The team quantified copy number aberrations in the samples and identified key patterns in the cell-free DNA recovered, confirming that fractions of cell-free DNA derived from the tumors in each sample matched what they expected based on orthogonal technologies. Interestingly, they could recover many long DNA fragments originating from the tumors. All of this was done rapidly, with DNA characterization and analysis performed within 24 hours of sample collection. Again, this suggests potential utility in a clinical trial setting, where time to results is often important for understanding unexpected reactions to a candidate therapy.
In one final example, researchers at the Dana-Farber Cancer Institute developed an innovative pipeline for single-cell transcriptomics.5 Their approach relies on long-read sequencing data to analyze single-cell cDNA libraries, identifying features associated with cell lineages such as isoforms, fusion genes, T cell receptors, and more. They evaluated this workflow by successfully tracking subclones from acute myeloid leukemia samples over time and suggested that it can be used by scientists for sophisticated cellular genotyping, particularly for tumor and immune cells.
Stay on top of what’s next
When long-read sequencing technologies such as nanopore-based tools were first introduced a decade ago, differences in the error profile of their sequence data raised concerns that these platforms might not be reliable enough for drug discovery and development programs. However, extensive independent benchmarking studies such as the PrecisionFDA Truth Challenge have shown that accuracy has now improved to the levels needed for even the most rigorous investigations on par with short-read technologies while adding structural variants and epigenomic profiles at the same time with no additional cost.6-8
With this increased accuracy and reliability, newer NGS tools are now ready for scientists in pharmaceutical and biotech companies. These technologies enable a more comprehensive view of DNA, RNA, and methylation, revealing clinically relevant elements missed by other sequencing platforms. The novel information they provide could fuel new avenues of research throughout drug discovery and development.
References
- Sergey Nurk et al. The complete sequence of a human genome. Science 376, 44-53 (2022). DOI: 10.1126/science.abj6987
- Beyter, D., Ingimundardottir, H., Oddsson, A. et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 53, 779–786 (2021). https://doi.org/10.1038/s41588-021-00865-4
- Lau, B.T., Almeda, A., Schauer, M. et al. Single-molecule methylation profiles of cell-free DNA in cancer with nanopore sequencing. Genome Med 15, 33 (2023). https://doi.org/10.1186/s13073-023-01178-3
- van der Pol Y, Tantyo NA, Evander N, Hentschel AE, et al. Real-time analysis of the cancer genome and fragmentome from plasma and urine cell-free DNA using nanopore sequencing. EMBO Mol Med. 2023 Dec 7;15(12):e17282. doi: 10.15252/emmm.202217282.
- Penter, L., Borji, M., Nagler, A. et al. Integrative genotyping of cancer and immune phenotypes by long-read sequencing. Nat Commun 15, 32 (2024). https://doi.org/10.1038/s41467-023-44137-7
- Olson ND, Wagner J, McDaniel J, Stephens SH, et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom. 2022 May 11;2(5):100129. doi: 10.1016/j.xgen.2022.100129.
- Ni Y, Liu X, Simeneh ZM, Yang M, Li R. Benchmarking of Nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput Struct Biotechnol J. 2023 Mar 24;21:2352-2364. doi: 10.1016/j.csbj.2023.03.038.
- Kolmogorov, M., Billingsley, K.J., Mastoras, M. et al. Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat Methods 20, 1483–1492 (2023). https://doi.org/10.1038/s41592-023-01993-x