Next-Generation Sequencing (NGS) technologies and techniques have improved the productivity and accuracy of DNA sequencers. The benefits of Moore’s Law are reducing the cost of the High Performance Computing (HPC) capabilities needed to perform the alignment and other workflow steps on the outputs from NGS sequencing machines. These two trends deliver tremendous benefit to advancing personalized medicine, pharmaceutical discovery, and innovations in life science research by continually lowering the overall cost of genomics.
The HPC systems for aligning and analyzing genome datasets comprise computational, storage, and fabric components—each of which can have an impact on the performance of a genomics workflow and thus on the cost for alignment. To understand the impact of HPC components on genome sequencing, a team of Hewlett Packard Enterprise (HPE) engineers recently evaluated the performance of different HPC systems on genome mapping workflows by benchmarking several of their HPE ProLiant servers.
The team used the Burrows-Wheeler Aligner (bio-bwa.sourceforge.net/) algorithm in the BWA kit software available from github.com/lh3/bwa/tree/master/bwakit, to map two different sets of data. They chose a workload that their customers use and publicly available reference genomes and datasets for their study.
- Benchmark 1: 10,000,009 101bp sequences of synthetic data against the hs38a reference genome.
- Benchmark 2: Run ERR009171, from the 1000 Genomes Project Pilot 2, consisting of 18,120,339 read pairs, against the human reference genome Homo_sapiens_assembly38.
Benchmark 1 is a single-end read experiment. Benchmark 2 is a paired-end experiment. The differences are technical; however, paired-end reads are more time-consuming to perform than single-end reads. For more details, refer to The Columbia Genome Center at https://systemsbiology.columbia.edu/genome-sequencing-defining-your-experiment.
The BWA kit includes the aligner, bwa-postalt.js script for handling ALT contigs when present, and SAMtools for file conversion from SAM to BAM format. Both mapping of ALT contigs and file conversion are serial processes, compared to the highly parallelized aligner. The bwamem script to start the alignment determines the steps of the workflow for each dataset and reference, resulting in potentially different CPU profiles for the chosen workflow.
The evaluation team was interested in looking at the following characteristics:
- Overall system performance and processing profile for each workflow.
- Effect of different generations of CPU families on serial performance.
- Impact of processor speed and core counts on overall system performance (serial and parallel).
- Storage performance of a Network File System (NFS) using 10 Gbps Ethernet versus a parallel file system (PFS) based on Lustre* using Intel® Omni-Path Architecture (Intel® OPA) fabric.
For the benchmarks, two generations of HPE ProLiant two-socket servers (see the following table) were used.
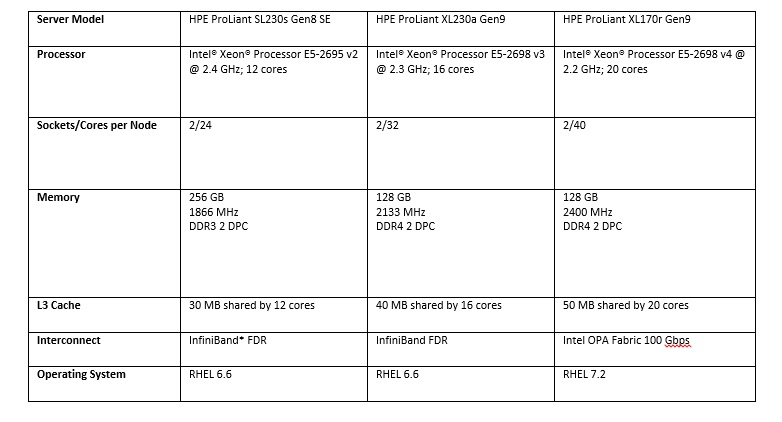
Workflows Matter
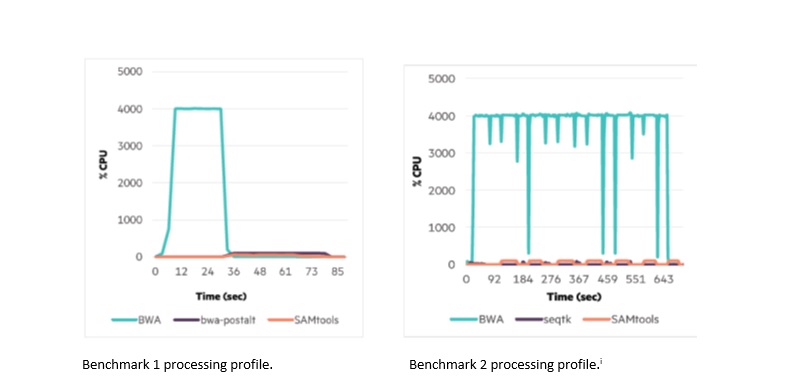
For the overall workflow performance, the team used the 40-core ProLiant XL170r Gen9 server with the 20-core Intel® Xeon® processor E5-2698 v4 and Intel Omni-Path Architecture fabric. This test revealed the variability in processing profiles, based on the workflow that bwamem chose for the datasets. (See the following figures.)
With benchmark 1, the parallelized aligner ran the entire dataset, consuming all 40 cores, in about one-third of the total mapping time. The process was followed by the serial bwa-postalt.js and SAMtools scripts, which used no more than one core each.
With benchmark 2, bwamem chose a different, much more parallel workflow by breaking up the read pairs into chunks. The script first ran seqtk to merge groups of the read pairs followed by the aligner, processing ALT contigs, and file conversion. The aligner was busy nearly the entire time, consuming all the cores, with short pauses for seqtk. In benchmark 2, the aligner competes for only two cores that bwa-postalt and SAMtools consume when they run. These two benchmarks convey that the workflow can significantly impact how the entire process utilizes the parallelism of the HPC system and the significance of serial tasks with workflows like benchmark 1.
CPU Models and Performance
Running the same workflows across the three servers with different generations of the Intel Xeon processor E5 family reveals how processor family affects performance, especially serial performance (see the following figure). Generally, serial performance is affected by processor and memory speeds. Different processor families support different memory, with the later processor generations taking advantage of faster memory. Benchmark 1, with its heavy serial component, benefits from about a 12 percent improvement from processor generation to generation, while benchmark 2 achieves about 8 percent improvement across generations. The indication here is that, with serial tasks, the later the processor generation, the more the workflow will benefit. But, how do core counts versus processor speed affect performance?
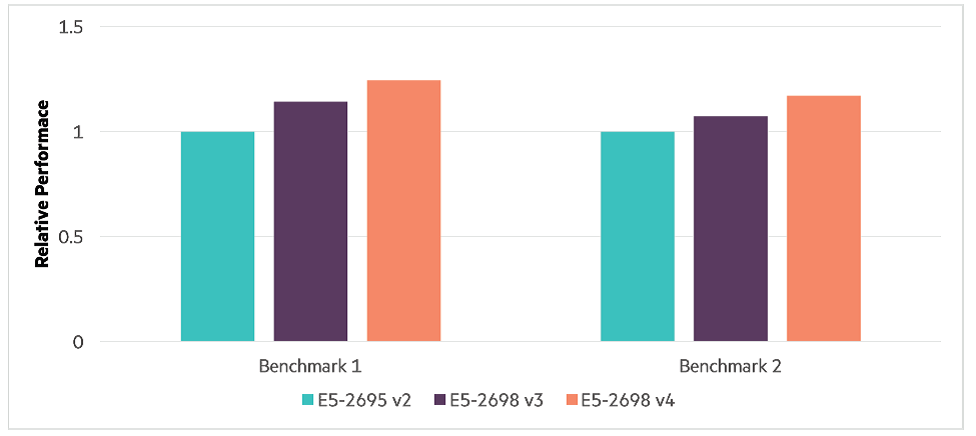
Performance due to processor families and memory.i
The HPE ProLiant XL170r Gen9 Server was repopulated with three different versions of the Intel® Xeon® processor E5 v4 family across the benchmarks:
- 14-core Intel® Xeon® processor E5-2690 v4 @ 2.6 GHz (28 cores per node);
- 20-core Intel® Xeon® processor E5-2698 v4 @ 2.2 GHz (40 cores per node); and
- 22-core Intel® Xeon® processor E5-2699 v4 @ 2.2 GHz (44 cores per node).
Benchmark 1 experiences a small benefit from the more cores, due to its dominant serial processes. But benchmark 2 jumps measurably with more cores available to the overall workflow—despite they are slower cores—which is not surprising considering its heavy parallelism.
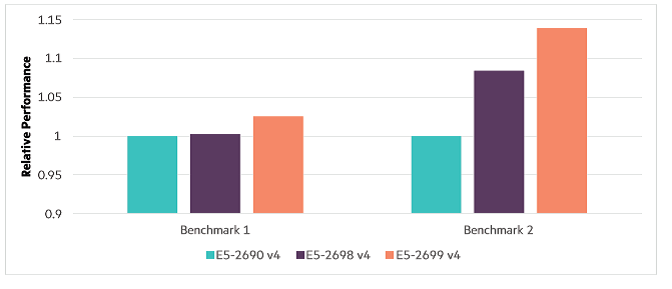
Performance from core count / processor speed.i
At many NGS centers, sequencers write data to a shared file system over a network, while the alignment and analysis nodes read the genomic data from the storage system. As the number of sequencers and compute nodes increases, it places growing stress on data reads and writes. So, the choice of storage system and interconnect has a significant impact on productivity in genomics and thus the cost per genome. NFS and Ethernet are often the shared file system and network of choice in many IT shops, whereas the Lustre parallel file systems is the leading storage system for the fastest HPC systems according to the latest TOP500 list (www.top500.org).
The team benchmarked the impact of the choice of storage and fabric on genome processing, by comparing the performance of NFS on Ethernet to Lustre on Intel Omni-Path Architecture fabric as the number of the 40-core nodes increased. (See the following table for systems used.)
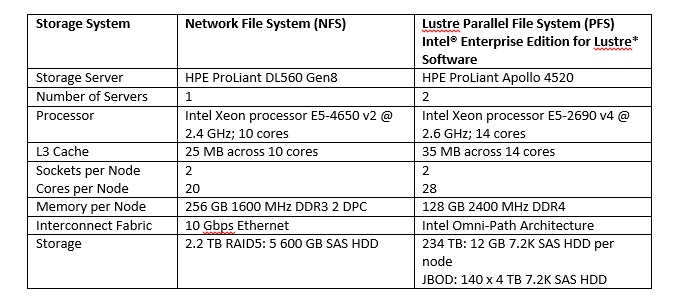
For both benchmarks, after eight nodes began accessing the NFS system, productivity decreased. Benchmark 2 experienced a more significant impact than Benchmark 1, with increasing degradation out to the 32 nodes used in the benchmark. Alternatively, Intel® Enterprise Edition for Lustre* software on the Intel OPA fabric continued to serve data to all nodes without a drop in productivity.
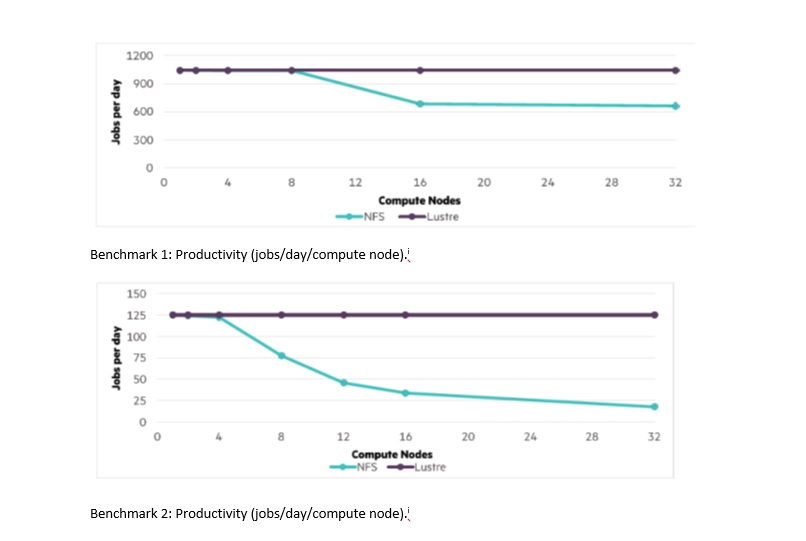
Lustre is widely used in the fastest HPC systems in the world because of its sustainable performance while supporting a very large number of nodes. Systems using Lustre often have many thousands of nodes, so the HPE team expects there remains plenty of headroom to support many more NGS sequencers beyond the 32 tested here.
Conclusions—Next-generation Technologies Improve Productivity in Genome Sequencing
NGS requires HPC solutions and storage infrastructures that deliver unmatched levels of performance, scalability, and affordability to support today’s genomics data-intensive workloads. With the datasets and workflows the HPE team tested, performance and productivity were highest with the latest generation of Intel Xeon processor E5 family, Intel Enterprise Edition for Lustre software, and Intel OPA fabric. Furthermore, the benchmarks revealed the following:
- The amount of parallelism in different workflows varies, impacting the CPU utilization profile.
- More cores, not higher processor frequency, deliver the best overall benefit.
- The Lustre parallel file system and Intel OPA fabric offer high performance with scalability.
The benchmarks also revealed other characteristics worth studying. The results are presented in the white paper “Accelerating Next-Generation Sequencing Workloads” available at https://www.hpe.com/h20195/v2/GetPDF.aspx/4AA6-6224ENW.pdf.
The cost of genome sequencing has a significant impact on the use of genomics to advance healthcare. As the performance and productivity of HPC goes up, the cost of sequencing genomes goes down, making it more readily available to more healthcare researchers and providers.
HPE’s study is only the beginning of further testing the company will complete to examine the efficiencies, performance, and cost benefits of advanced computing technologies for NGS.
[1] Used with permission from Hewlett Packard Enterprise



