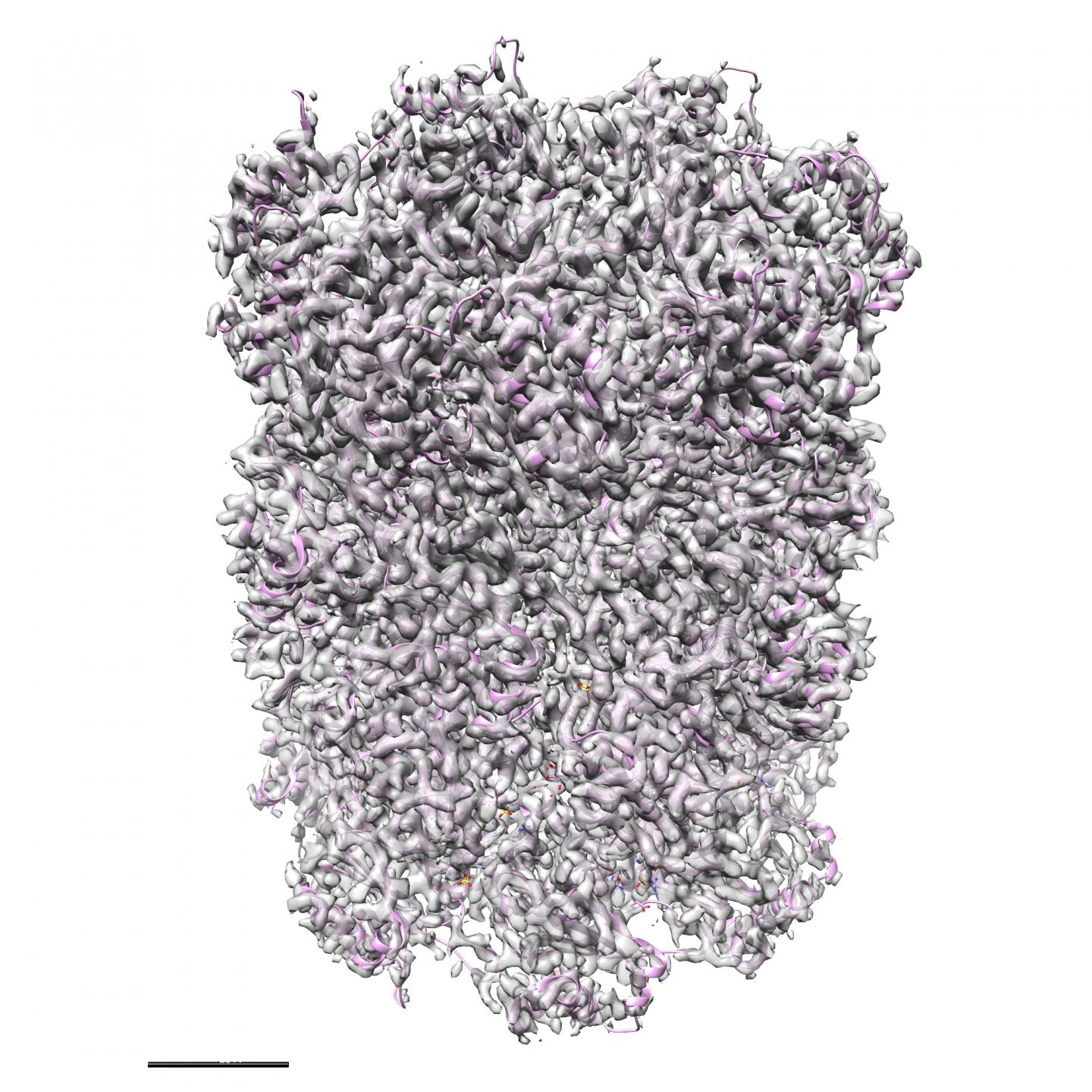
A new set of machine learning algorithms developed at U of T Scarborough that can generate 3-D structures of tiny protein molecules may revolutionize the development of drug therapies for a range of diseases. Source: Structura Biotechnology Inc
A new set of machine learning algorithms developed by U of T researchers that can generate 3D structures of tiny protein molecules may revolutionize the development of drug therapies for a range of diseases, from Alzheimer’s to cancer.
“Designing successful drugs is like solving a puzzle,” says U of T PhD student Ali Punjani, who helped develop the algorithms.
“Without knowing the three-dimensional shape of a protein, it would be like trying to solve that puzzle with a blindfold on.”
The ability to determine the 3D atomic structure of protein molecules is critical in understanding how they work and how they will respond to drug therapies, notes Punjani.
Drugs work by binding to a specific protein molecule and changing its 3D shape, altering the way it works once inside the body. The ideal drug is designed in a shape that will only bind to a specific protein or proteins involved in a disease while eliminating side effects that occur when drugs bind to other proteins in the body.
This new set of algorithms reconstructs 3D structures of protein molecules using microscopic images. Since proteins are tiny — even smaller than a wavelength of light – they can’t be seen directly without using sophisticated techniques like electron cryomicroscopy (cryo-EM). This new method is revolutionizing the way scientists can discover 3D protein structures, allowing the study of many proteins that simply could not be studied in the past.
Cryo-EM is unique because it uses high-power microscopes to take tens of thousands of low-resolution images of a frozen protein sample from different positions. The computational problem is to then piece together the correct high-resolution 3D structure from the low-resolution 2D images.
“Our approach solves some of the major problems in terms of speed and number of structures you can determine,” says Professor David Fleet, chair of the Computer and Mathematical Sciences Department at U of T Scarborough and Punjani’s PhD supervisor.
The algorithms, which were co-developed by Fleet’s former Post-Doctoral Fellow Marcus Brubaker, now an Assistant Professor at York University, could significantly aid in the development of new drugs because they provide a faster, more efficient means at arriving at the correct protein structure.
“Existing techniques take several days or even weeks to generate a 3D structure on a cluster of computers,” says Brubaker. “Our approach can make it possible in minutes on a single computer.”
Punjani adds that existing techniques often generate incorrect structures unless the user provides an accurate guess of the molecule being studied. What’s novel about their approach is that it eliminates the need for prior knowledge about the protein molecule being studied.
“We hope this will allow discoveries to happen at a ground-breaking pace in structural biology,” says Punjani. “The ultimate goal is that it will directly lead to new drug candidates for diseases, and a much deeper understanding of how life works at the atomic level.”